
Abstract
Objective: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that is incurable and ultimately fatal. Few therapeutic options are available to patients. In this study, we explored differences in microbiome composition associated with ALS. Methods: We compared the gut microbiome and inflammatory marker profiles of ALS patients (n = 10) to those of their spouses (n = 10). Gut microbiome profiles were determined by 16S rRNA gene sequencing. Results: The gut microbial communities of the ALS patients were more diverse and were deficient in Prevotella spp. compared with those of their spouses. In contrast, healthy couples (n = 10 couples of the opposite sex) recruited from the same geographic region as the patient population did not exhibit these differences. Stool and plasma inflammatory markers were similar between ALS patients and their spouses. Predictive analysis of microbial enzymes revealed that ALS patients had decreased activity in several metabolic pathways, including carbon metabolism, butyrate metabolism, and systems involving histidine kinase and response regulators. Conclusions: ALS patients exhibit differences in their gut microbial communities compared with spouse controls. Our findings suggest that modifying the gut microbiome, such as via amelioration of Prevotella spp. deficiency, and/or altering butyrate metabolism may have translational value for ALS treatment.
Introduction
Studies using animal models of amyotrophic lateral sclerosis (ALS) suggest that gut microbiome dysregulation may play a role in ALS pathophysiology. The murine ALS model that expresses a mutant form of human superoxide dismutase 1 (SOD1G93A) exhibits damaged gut epithelial cell tight junctions, increased gut permeability, and shifted gut microbiome profiles compared with wild-type mice (Citation1). In particular, these animals have proportionately fewer microbes that produce butyrate, a short-chain fatty acid. Butyrate dietary supplementation appears to be protective and lengthens both the onset time of weight loss and survival in these mice (Citation2). Moreover, reductions in immune-stimulating bacteria appear to be protective from premature mortality in a c9orf72-mutant mouse model (Citation3). However, evidence regarding the gut microbiome in humans with ALS is less clear.
A small study reported altered gut microbiome profiles in ALS patients (n = 6), again with proportionately fewer butyrate-producing microbes, compared with healthy controls (n = 5) (Citation4). Another study found that ALS patients (n = 5) had a less diverse microbiota than healthy controls, but the source and number of controls were not specified (Citation5). Most recently, a large study of 66 people with ALS and 61 healthy controls found that the relative abundance of butyrate-producing bacteria was significantly lower in ALS patients compared to healthy controls (Citation6). In contrast, a larger study showed that ALS patients (n = 25) exhibited higher microbiome species richness, as quantified by 16S rRNA gene amplicon sequence variants (ASVs), indicating a higher number of total species compared with age- and sex-matched controls. However, there was no difference in species evenness, as indicated by the Shannon and Simpson’s indices (Citation7). Additionally, ALS patients had a significantly higher proportion of sequences from the Ruminococcaceae family (genus and species unknown), but no other differences in the relative composition of the microbiome were observed compared with the controls (Citation7). Similarly, no differences associated with functional predictions were observed (Citation7). Mazzini et al. (Citation8) also compared ALS patients with age- and sex-matched healthy controls (n = 50) and found that the ALS patients had a higher abundance of Escherichia coli and Enterobacter spp. as well as a lower abundance of yeasts. The apparent contradictions of these studies may be due to differences in the control groups (age- and sex-matched (Citation7,Citation8) vs. unmatched), sequencing technology (pyrosequencing (Citation4,Citation7) vs. PCR (Citation5,Citation8)) and platforms, bacterial genome sequencing targets (V3-V4 region of the 16S rRNA gene (Citation4) vs. V2-V3 region of the 16S rRNA gene and D1 region of the 26S rRNA gene (Citation8)), reference databases (Silva123 (Citation7), NCBI (Citation5), or unspecified), and bioinformatics software (QIIME v1 (Citation4), ClustalW2 (Citation5), or unspecified) used. Furthermore, various methodological differences among these previous human ALS studies prevent an inference of stronger evidence supporting one study’s findings over the others.
Another study examined the gut microbiota in both the ALS mouse model and human ALS patients (Citation9). SOD1G93A mice exhibited gut microbial dysbiosis that was exacerbated after antibiotic administration. Symptom severity was associated with 11 distinct strains of bacteria. Of these, Akkermansia muciniphila ameliorated ALS symptoms, whereas Ruminococcus torques and Parabacteroides distasonis exacerbated them. A. muciniphila administration resulted in central nervous system nicotinamide accumulation, and nicotinamide dietary supplementation improved motor symptoms in the mice. Similarly, ALS patients (n = 37) had different microbiome compositions and lower levels of nicotinamide compared with body mass index- and age-matched family members (n = 29).
Here, we examined the gut microbiota of ALS patients using a novel study design in which the patients’ spouses served as respective controls. Several reports have established gut microbial composition similarities among cohabitating people due to environment and diet commonalities. This has been particularly observed with physically intimate partners (Citation10–13). Thus, the spouses of the ALS patients provided a natural control group, thus establishing the null hypothesis of a lack of difference in gut microbial composition and effectively excluding any differences due to environment, diet, and physical intimacy. We conducted comparisons between the ALS patients and their spouses with respect to gut microbiome compositional profiles and a comprehensive set of stool and plasma inflammatory biomarkers.
Materials and methods
Participants and setting
A total of 40 participants were recruited into this study. The “ALS group” consisted of 20 individuals, 10 patients who were recruited from the ALS Center of Emory University, and their spouses were recruited as controls. The “healthy volunteer group” included 10 healthy couples (20 individuals) who volunteered to serve as geographical controls. Inclusion criteria included: (1) patients with live-in spouses who were willing and eligible to participate, and (2) participants between the age of 18–79 years. Patients receiving enteral nutrition as well as those with a history of bowel disease other than constipation, malignancy, dementia/other cognitive disorders, or Parkinson’s disease (PD)/other neurodegenerative diseases were excluded. The study was approved by the Emory University Institutional Review Board (IRB00078771).
Participant recruitment
Patients diagnosed with ALS were identified by research staff at the Emory ALS Center, who reviewed the lists of individuals with clinic appointments over the 4 months in which we recruited. Research staff sought to identify patients who were within 2 years of symptom onset and who had spouse caregivers. About one-third of eligible couples were not approached due to lack of research staff for this unfunded study, and approximately half of couples who were approached agreed to participate. Most of the prospective participants who declined did so because of concerns about the rectal swab or blood draw.
Biological sample collection
One rectal swab was obtained from each participant in the healthy volunteer group, and two rectal swabs were obtained from each participant in the ALS group. Each participant received verbal and pictorial instructions on how to easily obtain a rectal swab using Sterile Catch-All Sample Collection swabs (Epicentre Biotechnologies, Madison, WI) with little or no discomfort. Swabs were stored in MoBio garnet bead tubes (MoBio Laboratories, Inc., Carlsbad, CA) and frozen upright on dry ice prior to transport to the laboratory. They were then stored at −80 °C until shipment on dry ice to the J. Craig Venter Institute in La Jolla, CA. The second swabs obtained from the ALS group participants were sealed in Parafilm and stored at room temperature prior to processing to determine stool inflammatory marker levels. In addition, the research coordinator collected blood samples via venipuncture from the ALS group participants, which were aliquoted into EDTA-treated tubes and frozen at −80 °C.
16s rRNA gene sequencing
DNA was extracted using the Qiagen DNeasy PowerSoil Kit, according to the manufacturer’s protocol (Catalogue # 12888-100; Qiagen, Inc. Germantown, MD). Using approximately 100 ng of sample DNA, the V4 variable region of the 16S rRNA gene was amplified by PCR using adaptor- and barcode-ligated primers (Citation14). The 515-533F forward (GTGCCAGCMGCCGCGGTAA) and 806-787R reverse (GGACTACHVGGGTWTCTAAT) primers were used. Library amplicons were purified using the QIAquick PCR Purification Kit (Qiagen, Inc.) and then quantified using the SYBRGold TECAN assay (Thermo-Fisher, Inc., Waltham, MA). Quantified libraries were normalized, pooled, and sequenced at a loading density of 10 pM on the Illumina MiSeq using the MiSeq Reagent kit v2 500 cycles supplemented with 20% PhiX, according to the manufacturer’s specifications. Mock community positive controls and negative quality control samples were processed in the same batch as the participant samples. Standard Illumina post-sequencing quality control software (cassava 1.8) was used to bin sequences according to dual indices and to remove poor quality sequences (>Q30) prior to data analysis.
Inflammatory biomarker analysis
Inflammatory biomarkers were measured in stool and plasma samples. Plasma supernatants were aliquoted and batch analyzed. The rectal swabs that were stored at room temperature were placed in 1 mL phosphate-buffered saline (PBS [pH 7.4]), and the stool was extracted using Qiagen TissueLyser at 4 °C. After centrifugation at 260 ×g for 10 min to clear debris, supernatants were aliquoted and batch analyzed. Plasma and stool samples were transported to the Emory Multiplexed Immunoassay Core for processing. Lipopolysaccharide (LPS) was measured in plasma using an enzyme-linked immunosorbent assay (ELISA) kit (Pierce LAL Chromogenic Endotoxin Quantitation Kit, Thermo-Fisher Scientific, Inc., Waltham, MA). Cytokines (interleukin [IL]-1β, IL-2, IL-4 IL-6, IL-8, IL-10, IL-12p70, IL-13, tumor necrosis factor [TNF], and interferon [IFN]-γ) were measured in stool and plasma samples using a V-PLEX Pro-inflammatory Panel 1 Human Kit in a 96-well plate on a QuickPlex instrument (Meso Scale Diagnostics, Rockville, MD). LPS-binding protein (LBP) and high-sensitivity C-reactive protein (hsCRP) were measured in stool and plasma samples using the Meso Scale Human LBP and CRP plates, respectively. All samples were processed in duplicate.
Bioinformatics processing
ASVs were generated from raw Illumina sequence reads using UPARSE (Citation15) and mothur (Citation16) open-source bioinformatics tools. Through this pipeline, paired-end reads were trimmed of adaptor sequences, barcodes, and primers, after which low-quality reads were discarded. After a de-replication step, sequences were filtered for chimeras using the UCHIME2 tool from UPARSE (Citation15,Citation17). Next, sequences were clustered with UPARSE set at 100% identity, thus producing ASVs (Citation18). We used mothur to predict taxonomy using the Wang classifier, bootstrapping 100 iterations. We reported taxonomies only when 80 or more of the 100 iterations were identical. Taxonomies were assigned to ASVs with mothur, using version 123 of the SILVA 16S ribosomal RNA database as the reference database (Citation19). We used DESeq2 to test for differential community composition (Citation20,Citation21).
Microbial community analyses
Microbial community analyses were performed using R statistical software (Citation22). To avoid the inclusion of possible sequencing artifacts, ASVs that were either found in fewer than five individuals or fewer than five times in the overall dataset were excluded from further analyses. Principal coordinate analyses were performed on Bray-Curtis distance matrices after transforming raw counts into proportions using the ape (Citation23) and vegan (Citation24) packages. The vegan package was used to calculate alpha diversity.
Statistical analyses
Group identity (ALS patient or spouse control) was not provided for initial group comparisons, but the pairing was preserved (i.e. the only group identity was A or B). To prevent unblinding, no demographic data were released during this initial analysis. Alpha diversity index comparisons between patients and their spouses were performed using the Student’s paired t-test or Wilcoxon signed-rank test, as appropriate. Group identities were revealed only after preliminary comparisons regarding alpha and beta diversity and relative ASV abundances were agreed upon. Principal coordinate analyses were also performed for the ALS group (ALS patients vs. spouse controls) and healthy volunteer group (males vs. females). We applied a Wald test (Citation25) to test for differential community composition. We used the Benjamini-Hochberg correction (Citation26) for multiple testing to identify genera, species, and inflammatory markers that differed between ALS patients and their spouse controls.
Predicted function comparisons
We used the SILVAngs pipeline (Citation19) with tax4fun (Citation27) to predict microbial enzyme abundance in the ALS patient and control samples. We performed t-tests to determine predicted differential enzyme abundances between the two groups and then selected enzymes with p values ≤0.05. The selected enzymes from each group were mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database to obtain functional profiles of each group.
Software
R version 3.6.2 and phyloseq version 1.30.0 were used to conduct statistical analyses and generate figures. Additional packages used are listed in the supplementary information.
Results
Ten pairs of patients (mean age: 50.8 ± 12.9 years, 70% male, 100% Caucasian, mean disease duration: 1.7 ± 1.1 years, mean ALS functional rating scale score—revised: 39.9 ± 3.6 points) and their opposite sex spouses (mean age: 47.5 ± 14.2 years, 30% male, 90% Caucasian) were enrolled. All ALS patients had a history of sporadic disease and negative testing for the C9orf72 mutation, the most common gene associated with ALS. No patients were taking riluzole, the most common treatment for ALS patients that can prolong survival for 2–3 months. None of the spouse controls had a history of chronic disease. The healthy volunteer group included 10 opposite sex couples (mean age: males, 47.2 ± 11.7 years; females, 49.4 ± 14.0 years). All were Caucasian. There were no significant differences with respect to age and race among the four groups: ALS patients, their spouses, and the males and females of the healthy volunteer group.
One patient/spouse pair was excluded from further 16S rRNA gene ASV analysis due to a lower number of reads from the patient sample (<1000) compared with the others. Rectal swab gut microbiome profiles from the nine ALS patients and their spouses and the 10 healthy control couples revealed 952 unique ASVs, clustered at 100% 16S rRNA gene sequence similarity (average number of reads per participant: 11,263). ASVs that were not detected more than three times in at least 20% of the samples across the 38 participants were eliminated, and 143 ASVs remained. These ASVs clustered into 85 genera and were used for further analyses.
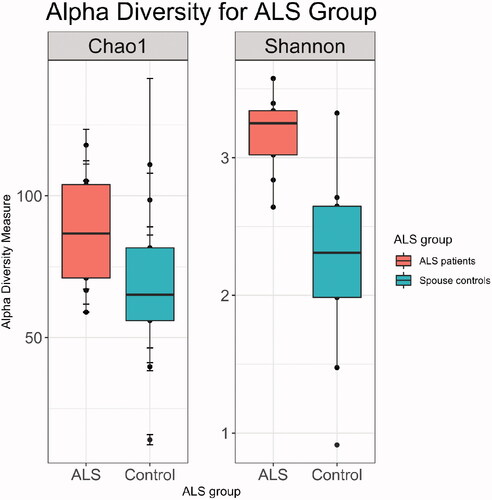
Principal coordinate analysis showed no differences in beta diversity between male and female spouses of the healthy volunteer group (by PERMANOVA: p = 0.362 for Bray-Curtis distance; p = 0.429 for unweighted UniFrac distance; p = 0.304 for weighted UniFrac distance) (). However, ALS patients differed from their spouses with respect to beta diversity (by PERMANOVA; p = 0.002 for Bray-Curtis distance; p = 0.107 by unweighted UniFrac distance; p = 0.004 for weighted UniFrac distance) (). Group centroids with the labeled pairs were plotted for the healthy volunteer (Supplementary Figure S1) and ALS (Supplementary Figure S2) groups. ALS patients had higher species richness and evenness than their spouses (Chao1 diversity: p = 0.03; Shannon diversity: p = 0.004; Wilcoxon signed-rank test; ).
Figure 1 Gut microbiome profiles did not differ between the males (n = 10) and females (n = 10) of the healthy volunteer group, as assessed by permutational analysis of variance (p = 0.4). PCoA: Principal coordinate analysis.
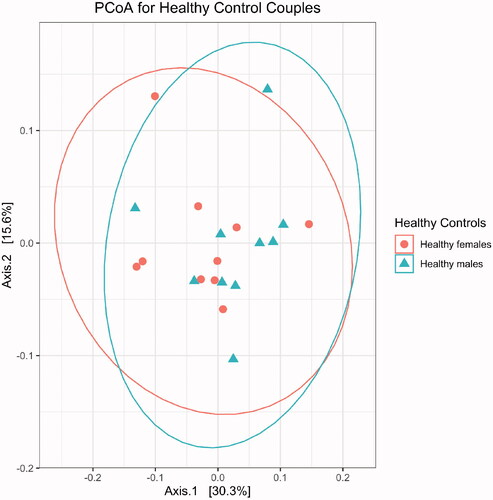
Figure 2 Gut microbiome profiles were different between ALS patients (n = 9) and their spouses (n = 9), as assessed by permutational analysis of variance (p = 0.003).
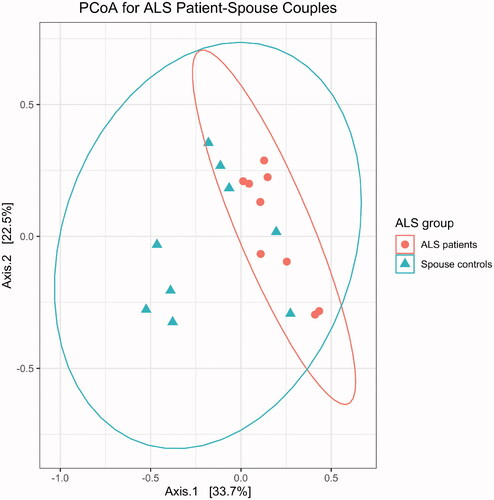
Figure 3 ALS patient (n = 9) gut microbiomes were more diverse than those of their spouses (n = 9) with respect to species richness (p = 0.03) and species evenness (p = 0.004), which were measured using the Chao1 and Shannon indices, respectively.
Compared with their spouses, ALS patients lacked ASVs of the genus Prevotella (adjusted p = 0.002; ). A particularly disparate ASV from the genus Prevotella in the two groups was P. timonensis (adjusted p = 9 × 10−18; ). However, none of the 25 comparisons of stool and plasma inflammatory biomarkers were statistically significant after correcting for multiple comparisons ().
Figure 4 ALS patients (n = 9) were deficient in Prevotella spp. compared with their spouses (n = 9; p = 0.02).
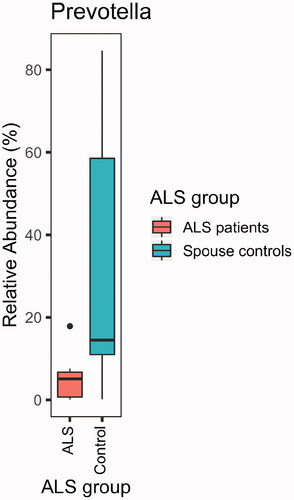
Figure 5 ALS patients (n = 9) were deficient in P. timonensis compared with their spouses (n = 9; p = 0.04; sign test).
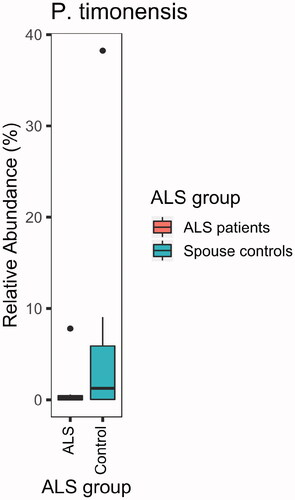
Table 1. Average (Avg.) and standard deviation (SD) values of inflammatory biomarkers in plasma and stool for ALS patients as well as for partner/caregiver controls, and the average and standard deviation of the difference between patient and control.
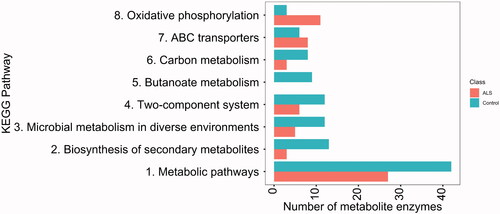
Predicted functional profile differences between the microbiomes of ALS patients and their spouses were determined (). The gut microbiomes of ALS patients exhibited lower levels of enzymes involved in some metabolic pathways, including carbon metabolism and systems involving histidine kinase and response regulators. Additionally, ALS patients completely lacked butanoate metabolic pathway enzymes, essential for butyrate production.
Figure 6 KEGG pathway analysis revealed a differential abundance of enzymes in ALS patients (upper bar) and their spouses (lower bar).
Discussion
In this study, we found differences in the gut microbiomes of ALS patients compared with their healthy spouses. ALS patient microbiomes were significantly more diverse with a higher number of taxa. ALS patients were also deficient in Prevotella spp. These differences were not found between couples in the healthy volunteer group.
Several human studies have previously reported an association between the gut microbiome and ALS pathophysiology (Citation3–6,Citation8); however, another study has suggested the contrary (Citation7). Gut microbiome differences have been described for several diseases, including irritable bowel syndrome (Citation28), psoriatic arthritis (Citation29), celiac disease (Citation30), atopic dermatitis (Citation31), Crohn’s disease (Citation32), and obesity (Citation33). In each case (Citation28–33), diseased patient microbiome profiles had lower alpha diversity (species richness and evenness) than those of controls, suggesting that diversity indicates a healthy gut. Thus, our finding of increased diversity with ALS was surprising. This finding might be due to stress-related responses in the spouses (Citation34–36). However, the lack of differences in key inflammatory cytokines indicated that either stress was exculpatory or that patients and spouses were equally stressed, albeit for different reasons.
A few studies have investigated gut microbiome profiles in PD patients and their spouses. Like ALS, PD is a neurodegenerative disease. Hill-Burns et al. (Citation37) reported that PD patients had significantly altered abundances of Bifidobacteriaceae, Christensenellaceae, Lachnospiraceae, Lactobacillaceae, Pasteurellaceae, and Verrucomicrobiaceae compared with controls, including some patient spouses and other community controls. In this same cohort, a secondary multivariate analysis of 37 immune and angiogenic factors in stool samples from patient/spouse pairs revealed that PD patients exhibited significantly higher levels of IL-1α, IL-1β, CXCL8, and CRP than their spouses, after controlling for sex, body mass index, smoking history, and probiotic use (Citation38).
The implications from these findings are critical for microbiome study design to meet the unbiased standard for the validity of a null hypothesis of no difference. The optimal study design should incorporate a spouse control group whenever possible because of influential environmental factors shared between patient/spouse pairs. Standard epidemiological parameter matching, including sex, age, and race, is not enough. Regardless of the matching process rigor, non-cohabitating case/control pairs do not share these same factors, thus affecting the study. We do note that use of spouse controls heightens the possibility of overmatching. In addition, the ALS patient was male for the majority of our ALS patient-spouse couples. In larger studies, comparisons of couples in which the patient is male to those in which the patient is female may address this possibility.
Our finding of lower Prevotella spp. proportions in ALS patients was consistent with other gut microbiome studies regarding neurodegenerative diseases. Gut microbiome dysbiosis has been associated with PD (Citation37,Citation39–45), multiple sclerosis (Citation46–50), and autism spectrum disorder (Citation51,Citation52). Some reports have indicated Prevotellaceae deficiencies in PD (Citation42–44), multiple sclerosis (Citation46–49), and autism spectrum disorder (Citation51). Again, inconsistencies among conclusions regarding the association of disease and lower levels of Prevotella spp. may have resulted from differences in technologies or control groups.
Prevotella spp. predominance in the gut microbiome has been associated with dietary fiber consumption (Citation53). Additionally, it has been shown that P. timonensis induced strong pro-inflammatory immune responses via dendritic cells, inducing dendritic cell maturation and Th1 cell differentiation (Citation54). Thus, P. timonensis predominance may have contributed to the lack of difference in the pro-inflammatory cytokines in ALS patients compared with their spouses.
Our functional predictions revealed that ALS patients lacked microbial enzymes associated with butyrate metabolism compared with their spouses. However, a deficiency in butyrate-producing microbes, as reported by others (Citation4,Citation6), was not observed. Nevertheless, our finding of the predicted lack of enzymes associated with butyrate metabolism along with a previously reported finding that butyrate supplementation delayed disease progression in SOD1G93A mice and the finding of deficiency of the butyrate-producing microbes Eubacterium rectale and Roseburia intestinalis in ALS patients relative to healthy controls in a larger human ALS study strongly suggest an association of butyrate availability in the gut and disease progression. Further studies of the association of ALS with butyrate-producing microbes are warranted.
Although Brenner et al. (Citation7) found no microbiome compositional differences between ALS patients and age- and sex-matched controls, they did find that ALS patients had higher species richness. This was consistent with our current findings. Results of the initial diversity analysis came to the principal investigators (V.S.H. and J.D.G.), who were blinded to the group identity (i.e. labeled as group A or B). The finding of higher species richness in ALS patient microbiomes was concerning because it has been believed that a diverse microbial community is associated with health. However, despite a decade of research, the definition of a healthy microbiome remains unclear.
A key strength of our study was use of spouses as controls. Additionally, we used a second set of healthy volunteer couples. Limitations of the study included the small sample size, thus restricting our ability to investigate the relationship of differential microbial communities with ALS characteristics such as site of onset as well as our ability to determine differential expression of markers of inflammation in plasma and stool between ALS patients and spouse controls. Moreover, 70% of the ALS patients were male, so gut microbiome differences due to sex may have dominated the case/control comparison. However, given the clustering of all ALS patients seen in the principal coordinate analysis (), this conclusion seems unlikely. Another limitation is the lack of detailed diet information. Finally, because of the cross-sectional nature of our study we were unable to investigate the relationship of differential microbial communities with temporal ALS characteristics such as disease progression rates.
In this study, we presented evidence of an altered gut microbiome in ALS patients relative to their spouses, which may have a role in ALS pathophysiology. Our findings of higher microbial diversity, Prevotella spp. deficiency, and differential compositional profiles in ALS patients compared with their spouses warrant further investigation to understand the role of gut microbes in ALS pathophysiology. Shotgun or whole-metagenome sequencing is necessary to determine the species and strains of Prevotella that differ in ALS patients, and further exploration of short-chain fatty acids, including butyrate, is needed to better understand its role in ALS pathophysiology. Our findings may have implications for developing new therapeutics for ALS that target the gut microbiome.
Ethics approval
This study was approved by the Emory University Institutional Review Board (IRB00078771).
Informed consent
All participants provided written informed consent.
Author contributions
Conceptualization (V.S.H., M.G.T., and J.D.G.); clinical investigation (C.N.F., M.P., and J.D.G.); laboratory investigation (C.A.K. and M.G.T. [JCVI]); 16S rRNA gene sequencing (K.E.N.); stool and plasma cytokine measurement (M.G.T. [EU]); data curation and formal analysis (V.S.H. [lead], H.S., and A.M.); funding acquisition (V.S.H. and K.E.N.); software and visualization (H.S. [lead], A.M., and V.S.H.); writing—original draft (V.S.H.); and writing—review and editing (V.S.H. [lead], H.S., C.N.F., A.M., M.P., C.A.K., M.T., M.G.T., K.E.N., and J.D.G.).
Supplemental Material
Download MS Word (358.7 KB)Acknowledgements
The authors are grateful for the participation of the patients and their spouses in this study.
Declaration of interest
The authors declare no competing interests.
Data availability statement
The 16S sequence data have been deposited in the National Center for Biotechnology Information (NCBI) database on BioProject accession number PRJNA566436. Sample metadata are maintained at dx.doi.org/10.15139/S3/84SGKO.
Code for processing and analyses are maintained at dx.doi.org/10.15139/53/845GKO.
Related Research Data
References
- Wu S, Yi J, Zhang Y, Zhou J, Sun J. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol Rep. 2015;3:e12356.
- Zhang YG, Wu S, Yi J, Xia Y, Jin D, Zhou J, et al. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin Ther. 2017;39:322–36.
- Burberry A, Wells MF, Limone F, Couto A, Smith KS, Keaney J, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020;582:89–94.
- Fang X, Wang X, Yang S, Meng F, Wang X, Wei H, et al. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front Microbiol. 2016;7:1479.
- Rowin J, Xia Y, Jung B, Sun J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol Rep. 2017;5:e13443.
- Nicholson K, Bjornevik K, Abu-Ali G, Chan J, Cortese M, Dedi B, et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020.
- Brenner D, Hiergeist A, Adis C, Mayer B, Gessner A, Ludolph AC, et al. The fecal microbiome of ALS patients. Neurobiol Aging. 2018;61:132–7.
- Mazzini L, Mogna L, De FM, Amoruso A, Pane M, Aloisio I, et al. Potential role of gut microbiota in ALS pathogenesis and possible novel therapeutic strategies. J Clin Gastroenterol. 2018;52:S68–S70.
- Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019;572:474–80.
- Brito IL, Gurry T, Zhao S, Huang K, Young SK, Shea TP, et al. Transmission of human-associated microbiota along family and social networks. Nat Microbiol. 2019;4:964–71.
- Dill-McFarland KA, Tang ZZ, Kemis JH, Kerby RL, Chen G, Palloni A, et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. 2019;9:703.
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–5.
- Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. Elife. 2013;2:e00458.
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20.
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
- Edgar R. UCHIME2: improved chimera prediction for amplicon sequencing. bioRxiv. 2016:074252.
- Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34:2371–5.
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
- Love M, Anders S, Huber W. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014;15:10.1186.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
- R Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2017.
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90.
- Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, et al. The vegan package. Community Ecology Package. 2007;10:631–7.
- Cule E, Vineis P, De Iorio M. Significance testing in ridge regression for genetic data. BMC Bioinformatics. 2011;12:372.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
- Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31:2882–4.
- Carroll IM, Ringel-Kulka T, Keku TO, Chang YH, Packey CD, Sartor RB, et al. Molecular analysis of the luminal- and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2011;301:G799–807.
- Scher JU, Ubeda C, Artacho A, Attur M, Isaac S, Reddy SM, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–39.
- Wacklin P, Kaukinen K, Tuovinen E, Collin P, Lindfors K, Partanen J, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis. 2013;19:934–41.
- Wang M, Karlsson C, Olsson C, Adlerberth I, Wold AE, Strachan DP, Martricardi PM, et al. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J Allergy Clin Immunol. 2008;121:129–34.
- Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–11.
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4.
- Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25:397–407.
- Knowles SR, Nelson EA, Palombo EA. Investigating the role of perceived stress on bacterial flora activity and salivary cortisol secretion: a possible mechanism underlying susceptibility to illness. Biol Psychol. 2008;77:132–7.
- O'Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65:263–7.
- Hill-Burns E, Debelius J, Morton J, Wissemann W, Lewis M, Wallen Z, et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord. 2017;32:739–49.
- Houser MC, Chang J, Factor SA, Molho ES, Zabetian CP, Hill‐Burns EM, et al. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov Disord. 2018;33:793–804.
- Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s Disease. PLoS One. 2015;10:e0142164.
- Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disord. 2015;30:1351–60.
- Sampson T, Debelius J, Thron T, Janssen S, Shastri G, Ilhan Z, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s Disease. Cell. 2016;167:1469–80.
- Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord. 2015;30:350–8.
- Unger MM, Spiegel J, Dillmann KU, Grundmann D, Philippeit H, Bürmann J, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66–72.
- Bedarf J, Hildebrand F, Coelho L, Sunagawa S, Bahram M, Goeser F, et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017;9:39.
- Hopfner F, Künstner A, Müller SH, Künzel S, Zeuner KE, Margraf NG, et al. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017;1667:41–5.
- Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Paz Soldan MM, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. 2016;6:28484.
- Cosorich I, Dalla-Costa G, Sorini C, Ferrarese R, Messina MJ, Dolpady J, et al. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv. 2017;3:e1700492.
- Jangi S, Gandhi R, Cox LM, Li N, von Glehn FAO, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015.
- Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the gut microbiota of patients with multiple aclerosis, with a striking depletion of species belonging to Clostridia XIVa and IV clusters. PLoS Biol. 2015;10:e0137429.
- Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. 2017;114:10713–8.
- De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, et al. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One. 2013;8:e76993.
- Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe. 2010;16:444–53.
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8.
- van Teijlingen NH, Helgers LC, Zijlstra-Willems EM, van Hamme JL, Ribeiro CM, Strijbis K, et al. Vaginal dysbiosis associated-bacteria Megasphaera elsdenii and Prevotella timonensis induce immune activation via dendritic cells. J Reprod Immunol. 2020;138:103085.