
Abstract
The kinesin family member 5A (KIF5A) motor domain variants are typically associated with hereditary spastic paraplegia (HSP) or Charcot-Marie-Tooth 2 (CMT2), while KIF5A tail variants predispose to amyotrophic lateral sclerosis (ALS) and neonatal intractable myoclonus. Variants within the stalk domain of KIF5A are relatively rare. We describe a family of three patients with a complex HSP phenotype and a likely pathogenic KIF5A stalk variant. More family members were reported to have walking difficulties. When reviewing the literature on KIF5A stalk variants, we found 22 other cases. The phenotypes varied with most cases having (complex) HSP/CMT2 or ALS. Symptom onset varied from childhood to adulthood and common additional symptoms for HSP are involvement of the upper limbs, sensorimotor polyneuropathy, and foot deformities. We conclude that KIF5A variants lead to a broad clinical spectrum of disease. Phenotype distribution according to variants in specific domains occurs often in the motor and tail domain but are not definite. However, variants in the stalk domain are not bound to a specific phenotype.
Introduction
The kinesin family member 5A (KIF5A) gene encodes for the neural kinesin heavy chain. It is part of the kinesin super family proteins (KIFs), that play an important role in intracellular transport and is expressed in neurons (Citation1–3). KIF5A contains a globular motor domain, also known as the “head”, an α-helical coiled-coil stalk domain and a globular “tail” domain. The head has an ATP-binding sequence and microtubule-binding sequence. It hydrolyzes ATP and transfers chemical energy, which results in movement of each KIF along microtubules, as a “rail” (Citation1,Citation4,Citation5). The stalk domain is used for kinesin dimerization and is important for interaction with other subunits and can bind to kinesin light chains, which are associated with certain cargos. Finally, the tail can directly bind to cargos and kinesin light chains. KIF5A plays a role in fast axonal transport and dendritic transport (Citation4,Citation5).
Variants in KIF5A are linked to a range of motor diseases depending on the location in the gene. For example, variants in the motor domain cause Charcot-Marie-Tooth disease 2 (CMT2) and spastic paraplegia type 10 (SPG 10, OMIM#604187) also known as hereditary spastic paraplegia (HSP), while variants in the cargo binding tail are associated with amyotrophic lateral sclerosis (ALS) (Citation3). Variants in the stalk domain are less well described.
HSP is a group of clinically and genetically heterogeneous neurodegenerative or neurodevelopmental disorders, which are characterized by lower limb spasticity and weakness (Citation6,Citation7). Based on the clinical phenotype it can be classified into pure or complex HSP. Patients with pure HSP predominantly show isolated pyramidal signs in the lower limbs but may also have sphincter dysfunction and sensory disturbances (vibration sense and joint position sense). Patients with complex HSP have more extensive neurological symptoms such as cognitive impairment, ataxia, polyneuropathy, epilepsy, skeletal abnormalities, upper limb amyotrophy, and optic atrophy (Citation6–8). SPG 10 is an autosomal dominant form of HSP, and most patients present with a complex HSP phenotype (Parkinsonism, cerebellar ataxia, cognitive impairment, axonal sensorimotor peripheral neuropathy, distal amyotrophy, pes cavus, and scoliosis). Childhood onset is most common (Citation7), but onset at adult age can also occur (Citation9).
CMT2 is characterized by chronic axonal motor and sensory polyneuropathy leading to progressive loss of sensation, weakness, muscle atrophy, loss of deep tendon reflexes, and foot deformities (Citation10).
ALS is a motor neuron disease (MND), characterized by combined degeneration of upper motor and lower motor neurons. Patients endure rapid progressive muscle weakness, atrophy, and spasticity, which may affect any voluntary muscle, making the clinical presentation and disease course very heterogeneous. On average, ALS patients develop respiratory failure within 3–4 years from onset, which is the most common cause of death. Currently, there is no effective treatment for ALS (Citation3,Citation11).
Finally, cases of a severe developmental syndrome, neonatal intractable myoclonus (NEIMY), have also been described due to de novo variants in the tail domain of KIF5A. The patients presented with myoclonic seizures and additional symptoms such as hypotonia, optic nerve abnormalities, dysphagia, apnea, early developmental arrest, and progressive leukoencephalopathy (Citation12,Citation13).
Here, we present the clinical and genetic findings in a pedigree with apparent autosomal dominant inheritance and a complex HSP phenotype due to a variant in the KIF5A stalk region. We subsequently performed a systematic search of the literature with the aim of providing an overview of genotype-phenotype correlations in KIF5A stalk domain variants.
Materials and methods
All patients in the present case series were seen at the neuromuscular outpatient clinic of the University Medical Centre Utrecht. Consent was provided for the use of clinical data for research purposes. To further characterize the clinical phenotype of KIF5A stalk variants, we performed a systematic search of the literature (Supplement A).
Results
Proband
A 38-year-old man presented with a four year history of progressive weakness of the lower limbs. Over the course of a few months, he had developed an inability to lift his left leg. The initial neurological exam was apparently normal, and consequently he was suspected of a conversion disorder. He was treated at a rehabilitation clinic for over a year, however this treatment was unsuccessful and discontinued. Subsequently, he was not seen by any medical professional until approximately four years after onset when his right leg also became impaired. This prompted him to reengage medical care, and eventually he was referred to our clinic. He reported frequent cramps in his calves and muscles spasms in both legs. He also complained of painful burning sensations in the lower limbs, which had also started four years ago. Light touch was painful, and he no longer tolerated wearing shoes. He did not have any problems in his arms or with his speech and swallowing.
Neurological examination showed normal function of cranial nerves and no pseudobulbar reflexes. There was no weakness or sensory deficits in the upper limbs. However, reflexes in the arms were pathologically brisk bilaterally including positive Hoffman-Trömner reflexes. Examination of the lower limbs showed subtle generalized atrophy of the entire left leg and bilateral pes cavus with hammer toes. Both legs were hypertonic, with pathologically brisk reflexes, sustained ankle clonus and positive Babinski sign bilaterally. He also had distal dysesthesia and hyperalgesia with normal vibration and position sense.
Routine laboratory tests (full cell blood count, glucose, coper, liver, renal and thyroid functions), showed no abnormalities, except for a vitamin B12 deficiency (101 pmol/L, reference value 130–700)), for which supplementation was started. Unfortunately, this did not ameliorate any of his symptoms. Serology testing for infectious causes was negative (Borrelia, HIV-1/2, HTLV 1/2, and Lues). Further metabolic screening, including very long chain fatty acid and urinary bile alcohols was normal (ruling out adrenoleukodystrophy and cerebrotendionous xanthomatosis). Brain and spinal cord MRI showed no abnormalities and routine electrophysiological testing revealed loss of motor axons to the right foot (no compound muscle action potential response peroneal nerve (m. extensor digitorum brevis)), normal sensory responses (sensory nerve action potential amplitudes and velocity) and normal needle electromyography (EMG).
Based on the clinical findings and results from ancillary tests, the patient was suspected to have an HSP phenotype with neuropathy or a hereditary motor sensory neuropathy phenotype with upper motor neuron involvement.
Family history
The proband reported that his father had similar complaints with weakness and stiffness of the lower limbs. As a child his father had mild trouble walking and was unable to perform sports with his peers. Starting from the age of approximately 18 years he developed pain in his legs as well as weakness. When he was 22 years, he suddenly lost the ability to move his legs, from which he recovered over the course of a few months. Unfortunately, there are no detailed medical records on this episode. He was however diagnosed with M. Bechterew around that time and he subsequently assumed that all his complaints could be attributed to this disorder. Over the course of the following year, he developed a pes equinus on the left side, for which he underwent surgical treatment.
Around age of 60 years he developed numbness and painful sensations in both feet, which had subsequently progressed to the level of his knees. He further went on to also develop deafness.
On neurological examination, we did not see any abnormalities of the cranial nerves or pseudobulbar reflexes. There was a slight tremor present, and hypertonia with cogwheel rigidity in the upper limbs (left more than right). There was no weakness, atrophy or fasciculations. He had bilateral, distal hypesthesia (from the wrist down), with slightly decreased vibration sense and symmetrically low reflexes. There was severe hypertonia and spasticity of the legs. There was no atrophy or fasciculations. He had hypesthesia from the knees down bilaterally with decreased vibration sense.
The proband’s sister has a mild mental disability. At the age of 38 years, she developed a progressive movement disorder of arms and legs, characterized by hypermobility, followed by dystonia and hypertonia, with impaired sensibility of the legs up to the pelvis and hallux valgus. She was recently diagnosed with a small bowel neuroendocrine tumor, for which she will undergo appropriate oncologic treatment.
Both father and sister underwent a similar work-up to proband (extensive laboratory and metabolic investigations, imaging, and EMG studies), which did not reveal any abnormalities.
Finally, the proband’s son of 11 years old was described to be clumsy and to have an unsteady gait making it impossible for him to walk long distances. Neurological examination showed no abnormalities of the cranial nerves or upper limbs. His legs were somewhat hypertonic, with brisk patellar reflexes bilaterally, brisk left ankle jerk reflex and ankle clonus on the right side. He had maximal strength in his legs and coordination tests and gait appeared normal. The parents opted not to perform genetic testing on their son. Therefore, his KIF5A status is unknown. Nevertheless, there seems to be an early HSP phenotype.
Genetic analyses
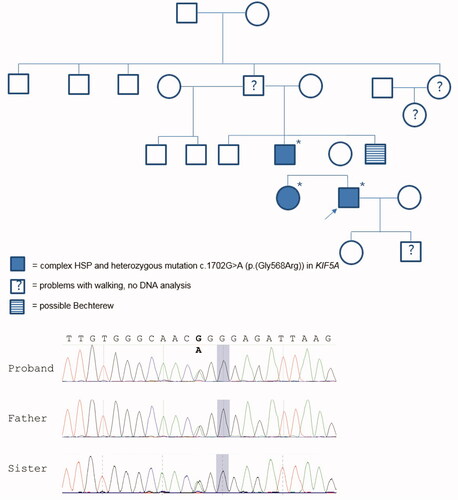
For the probound exome sequencing was performed through the Radboud University Medical Centre. Capture of exons was done using an Agilent SureSelect Human All Exon 50 Mb Kit (Santa Clara, CA) and sequencing was performed using Illumina HiSeq, both done by BGI-Europe (Copenhagen, Denmark). Median coverage depth was at least 80x. Subsequently, read mapping and variant calling were done using the Burrows-Wheeler Aligner (mapping) and GATK (calling) and copy number variant analyses were done using CoNIFER. The datasets were analyzed using an in-house annotation pipeline and manual interpretation by a clinical laboratory geneticist (Citation14). Finally a movement disorder gene panel containing 225 genes (Supplement B) was analyzed. This showed a heterozygous variant c.1702G > A p.(Gly568Arg) in the KIF5A gene. When his father and sister were tested (Sanger sequencing), it was revealed that they both were also heterozygous carriers of the same variant in KIF5A.
The probands variant was considered a variant of unknown significance (VUS) as the variant is not located in the kinesin motor domain. This variant has a low frequency of three in >125.000 persons in the gnomAD control population (Citation15). It affects an evolutionary conserved Glycine at this position in alignments of KIF5A orthologues (vertebrates and fruit fly) and the variant has a combined annotation dependent depletion (CADD) score of 22.6 (PHRED) (Citation16). The fact that his affected family members (father and sister) had the same variant and similar symptoms, made it more likely that the variant is pathogenic.
When going through the family history at greater depth, it became apparent that there are multiple additional family members who had problems with walking (see ). Medical records and/or DNA samples were not available for these individuals.
Figure 1 Pedigree of examined family including genotype/phenotype information. Females are represented by circles and males by squares. The proband is marked with an arrow and * indicates the cases that have a known genotype. Sequence chromatogram of proband, his father, and sister showing the c.1702G > A variant (bold).
Literature review
Our systematic search resulted in 146 articles. After screening title/abstract on KIF5A variants 48 articles remained that were screened on full text (Supplement A). Thirty-nine articles that mentioned KIF5A variants were found (reviews were excluded).
For the KIF5A protein the following regions were considered as a domain, amino acid 9 to 327 for the motor domain, amino acid 331 to 906 for the stalk domain, and amino acid 907 to 1.032 for the tail domain (Citation3).
We identified 22 cases with a KIF5A stalk variants published in the literature. Genetic data, clinical information, gnomAD allele count and frequency (Citation15), single nucleotide polymorphism database reference ID, the American College of Medical Genetics classification and CADD scores (Citation16) of the proband, probands father and the literature cases are summarized in . Reported KIF5A variants in the literature (all domains) and the associated phenotypes are illustrated in .
Figure 2 Localization of reported KIF5A variants within the different regions of the gene and the phenotypes associated with them. Variants with an allele frequency above 1/1000 in gnomAD v2.1.1 are in gray. ALS: amyotrophic lateral sclerosis; CMT2: Charcot-Marie-Tooth; NEIMY: neonatal intractable myoclonus; SPG: spastic paraplegia.
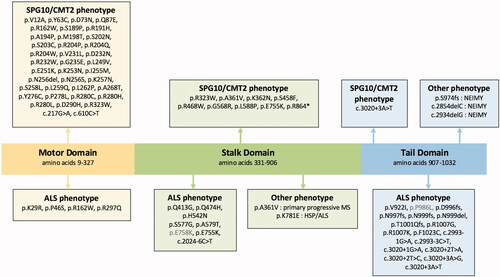
Table 1 Reported KIF5A stalk variants.
Discussion
The KIF5A gene contains three domains, the motor, stalk, and cargo binding domains, with clear genotype-phenotype correlations for variants in the motor domain (HSP/CMT2) and cargo binding domain (ALS). In contrast, stalk domain variants are relatively rare and are less well characterized.
We reported a family with a variant in the stalk domain of KIF5A, all presenting with a complex HSP phenotype. This included a variable onset (childhood-adult age) with weakness, hypertonia and spasticity of the legs, mainly distal sensory disturbances in the legs, hyperreflexia, and foot deformities. One of them also has an intellectual disability. This could be an additional symptom related to the syndrome but is not described in other cases of KIF5A stalk variants. In family members with an unknown genotype, difficulties with walking and possible Bechterew’s disease were also described.
When we reviewed the published cases with KIF5A stalk domain variants (), we found a broad phenotypic variability. Several cases of ALS were reported but most patients had a complex HSP phenotype. Involvement of the upper limbs, sensorimotor polyneuropathy, and foot deformities were reported most frequently. Also, the age of onset varied from early childhood to as late as the age of 54 years. The family in the present case-series also has this phenotype.
When studying the literature on variants within the motor domain, we found that these are associated with pure or complex forms of HSP (SPG10) and CMT2 (Citation17). Additional symptoms in these complex forms of HSP include axonal sensorimotor neuropathy (Citation18–32), foot deformities such as pes cavus (Citation19,Citation20,Citation22,Citation23,Citation30,Citation33,Citation34), tremor (Citation25), attention deficit hyperactivity disorder (Citation27), cognitive dysfunction (Citation28), behavioral changes (Citation33), ataxia (Citation21,Citation28,Citation34), autonomic dysfunction (Citation30), and congenital neurosensorial deafness (Citation20). Although onset is mainly seen during childhood, adult onset has also been reported in a few cases (Citation9,Citation20,Citation22,Citation25,Citation30,Citation33,Citation35).
In patients diagnosed with CMT2 due to a KIF5A motor domain variant, the phenotype is typically characterized by a motor and sensory polyneuropathy and pes cavus (Citation33). However, additional symptoms have also been reported and include spasticity and/or pyramidal signs (Citation28,Citation36,Citation37), cognitive dysfunction (Citation28,Citation37), ataxia (Citation36), and deafness (Citation37). Taken together, it appears that the phenotypic characteristics of the KIF5A motor domain variants all belong to a spectrum in which neuropathy with foot deformities and upper motor neuron signs are the main core features. The additional phenotypic characteristics (such as cognitive dysfunction (Citation28,Citation37), ataxia (Citation36), and deafness (Citation37)) also overlap. Therefore, the previous descriptions of CMT2 and HSP cases with motor domain variants could perhaps be best characterized as complex HSP.
Other phenotypes have also been described in patients with KIF5A motor domain variants, such as adult-onset distal spinal muscular atrophy (Citation38).
Variants in the tail domain are mainly associated with ALS (Citation3,Citation37,Citation39–41), although other phenotypes have been described, such as NEIMY (Citation12,Citation13). In one family whole exome sequencing identified a heterozygous variant, in exon 27 of KIF5A (tail domain), that segregated with disease, resulting in varying phenotypes such as spastic ataxia plus (nystagmus and hyperreflexia), cerebellar ataxia (with nystagmus), and MND plus cerebellar ataxia (Citation42).
Interestingly variants of unknown significance in the KIF5A motor domain have also been reported in patients with ALS (Citation37), as well as in a family in which patients presented with a late-onset SPG10 phenotype and later developed dysarthria and lower motor neuron signs with fasciculations (Citation23). Additionally, as shown in , 11 cases with a KIF5A stalk variant have an ALS-like phenotype. One patient presented with a HSP phenotype as a child and developed bulbar symptoms and lower motor neuron signs around the age of 14 years old. This patient had a heterozygous KIF5A stalk variant (paternal) and a homozygous ALS2 variant. The mother with only a heterozygous ALS2 variant was healthy, but the father with both a heterozygous KIF5A and ALS2 variant developed a slowly progressive ALS phenotype around the age of 50 years. The father had a brother that had ALS and died at the age of 50 years (Citation43). Since the mother was healthy and had a ALS2 variant, the KIF5A stalk variant is likely to play a role in the disease of the father. However, for the child it remains uncertain which variant plays which role in the course of disease. Another case of a KIF5A stalk variant associated with ALS was a patient, with a negative family history, described by Filosto et al. (Citation2). The patient presented with a slowly progressive motor syndrome that resembles ALS and HSP with an axonal neuropathy over the course of four years. Further course of disease is unfortunately unknown (Citation2). Finally, seven cases with KIF5A stalk variants and one case of a KIF5A stalk-and CCNF variant were found. According to the authors all these variances were of uncertain significance (Citation39,Citation41,Citation44).
The fact that an ALS phenotype is not limited to variants in the tail domain of KIF5A suggests that the phenotypic distribution according to variants in specific domains is not absolute. However, the cases described with a stalk variant seem to have a slower disease progression, so ALS phenotypes due to a KIF5A stalk variant might be relatively mild.
It can be challenging to determine the pathogenicity of rare variants found in single cases or families. A detailed family history to identify relatives with related phenotypes may be helpful. However, the feasibility of cosegregation analysis may be limited, especially in diseases with an adult onset, where older family members may already be deceased, and younger family members may carry the variant but not have symptoms (yet). This is of importance because false assignments of pathogenicity can have severe consequences for patients and their families (Citation45). In this article we aimed to create a full overview of KIF5A variants, so all reported KIF5A variants are mentioned, however we did differentiate between variants that have an allele frequency over 1/1000, since this is used as cutoff in the clinical practice to counsel family members. It is also important to mention that the pathogenicity of the stalk mutations is not definite, as all the mutations reported are a VUS.
Conclusion
Variants within different domains of KIF5A appear to result in a distinct neurological phenotype. However, these phenotypes are not necessarily limited to one domain and may even differ within family members sharing the same variant, resulting in a broad clinical spectrum due to KIF5A variants. This may range from a patient with HSP who has a father with only mildly brisk reflexes (Citation33), to a father developing HSP at the age of 30 years but having a child with a clumsy gait from the age of 8 years (Citation24) to a family with highly variable manifestations of complex HSP, cerebellar ataxia, and motor neuron disease (Citation42).
Moreover, variants in the stalk domain of KIF5A most often result in complex HSP phenotype but can also be associated with ALS.
Declaration of interest
The authors report no conflicts of interest regarding the contents of this manuscript.
JHV reports to have sponsored research agreements with Biogen.
LHvdB reports grants from ALS Foundation Netherlands, The Netherlands Organization for Health Research and Development (VICI scheme, funded through the EU Joint Programme-Neurodegenerative Disease Research, JPND [SOPHIA, STRENGTH, ALS-CarE projects), and personal fees from Shire, Cytokinetics, and Treeway, outside the submitted work.
MAvE received grants from the Netherlands Organization for Health Research and Development (VENI scheme), The Thierry Latran foundation and the Netherlands ALS foundation (Stichting ALS Nederland), the MND association, FIGHT-MND, and the EU Joint Programme-Neurodegenerative Disease Research (JPND), has consulted for Biogen and serves on the medical ethical review board of the UMC Utrecht.
References
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10: 682–96.
- Filosto M, Piccinelli S, Palmieri I, Necchini N, Valente M, Zanella I, et al. A novel mutation in the stalk domain of KIF5A causes a slowly progressive atypical motor syndrome. J Clin Microbiol. 2018;8:17.
- Nicolas A, Kenna KP, Renton AE, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2019;97:1268–83.e6.
- Hirokawa N, Noda Y. Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev. 2008;88:1089–118.
- Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–76.
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol. 2014;261:518–39.
- de Souza PVS, de Rezende Pinto WBV, de Rezende Batistella GN, Bortholin T, Oliveira ASB. Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum. 2017;16:525–51.
- Goizet C, Boukhris A, Mundwiller E, Tallaksen C, Forlani S, Toutain A, et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat. 2009;30:E376–85.
- Blair MA, Ma S, Hedera P. Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics 2006;7:47–50.
- Crimella C, Tonelli A, Airoldi G, Baschirotto C, D'Angelo MG, Bonato S, et al. The GST domain of GDAP1 is a frequent target of mutations in the dominant form of axonal Charcot Marie Tooth type 2K. J Med Genet. 2010;47:712–6.
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–98.
- Duis J, Dean S, Applegate C, Harper A, Xiao R, He W, et al. KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann Neurol. 2016;80:633–7.
- Rydzanicz M, Jagła M, Kosinska J, Tomasik T, Sobczak A, Pollak A, et al. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin Genet. 2017;91:769–73.
- Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg E-J, Mensenkamp AR, et al. A post-hoc comparison of the utility of Sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–6.
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43.
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, et al. A kinesin heavy chain KIF5A mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet. 2002;71:1189–94.
- Cuchanski M, Baldwin KJ. Mutation in KIF5A c.610C > T causing hereditary spastic paraplegia with axonal sensorimotor neuropathy. Case Rep Neurol. 2018;10:165–8.
- Musumeci O, Bassi MT, Mazzeo A, Grandis M, Crimella C, Martinuzzi A, et al. A novel mutation in KIF5A gene causing hereditary spastic paraplegia with axonal neuropathy. Neurol Sci. 2011;32:665–8.
- Muglia M, Citrigno L, D'Errico E, Magariello A, Distaso E, Gasparro AA, et al. A novel KIF5A mutation in an Italian family marked by spastic paraparesis and congenital deafness. J Neurol Sci. 2014;343:218–20.
- Qiu Y, Zhong S, Cong L, Xin L, Gao X, Zhang J, et al. A novel KIF5A gene variant causes spastic paraplegia and cerebellar ataxia. Ann Clin Transl Neurol. 2018;5:1415–20.
- Dohrn MF, Glöckle N, Mulahasanovic L, Heller C, Mohr J, Bauer C, et al. Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem. 2017;143:507–22.
- Kaji S, Kawarai T, Miyamoto R, Nodera H, Pedace L, Orlacchio A, et al. Late-onset spastic paraplegia type 10 (SPG10) family presenting with bulbar symptoms and fasciculations mimicking amyotrophic lateral sclerosis. J Neurol Sci. 2016;364:45–9.
- Lee H, La Y, Na HK, Kim H, Shin S, Choi YC. Hereditary spastic paraplegia with axonal sensorimotor polyneuropathy in a Korean family caused by pathogenic variant of KIF5A (c.611G > A). J Clin Neurol. 2020;16:347–8.
- López E, Casasnovas C, Giménez J, Santamaría R, Terrazas JM, Volpini V. Identification of two novel KIF5A mutations in hereditary spastic paraplegia associated with mild peripheral neuropathy. J Neurol Sci. 2015;358:422–7.
- Andréasson M, Lagerstedt-Robinson K, Samuelsson K, Solders G, Blennow K, Paucar M, et al. Altered CSF levels of monoamines in hereditary spastic paraparesis 10: A case series. Neurol Genet. 2019;5:e344.
- Jerath NU, Grider T, Shy ME. Progressive lower extremity weakness and axonal sensorimotor polyneuropathy from a mutation in KIF5A (c.611G > A;p.Arg204Gln). Case Rep Genet. 2015;2015:1–5.
- Liu Y-T, Laurá M, Hersheson J, Horga A, Jaunmuktane Z, Brandner S, et al. Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology 2014;83:612–9.
- Schüle R, Kremer BPH, Kassubek J, Auer-Grumbach M, Kostic V, Klopstock T, et al. SPG10 is a rare cause of spastic paraplegia in European families. J Neurol Neurosurg Psychiatry. 2008;79:584–7.
- Collongues N, Depienne C, Boehm N, Echaniz-Laguna A, Samama B, Dürr A, et al. Novel SPG10 mutation associated with dysautonomia, spinal cord atrophy, and skin biopsy abnormality. Eur J Neurol. 2013;20:398–401.
- Citrigno L, Magariello A, Pugliese P, Di Palma G, Conforti FL, Petrone A, et al. Kinesins in neurological inherited diseases: a novel motor-domain mutation in KIF5A gene in a patient from Southern Italy affected by hereditary spastic paraplegia. Acta Neurol Belg. 2018;118:643–6.
- Rinaldi F, Bassi MT, Todeschini A, Rota S, Arnoldi A, Padovani A, et al. A novel mutation in motor domain of KIF5A associated with an HSP/axonal neuropathy phenotype. J Clin Neuromuscul Dis. 2015;16:153–8.
- Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet. 2012;82:157–64.
- Morais S, Raymond L, Mairey M, Coutinho P, Brandão E, Ribeiro P, et al. Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur J Hum Genet. 2017;25:1217–28.
- Elert-Dobkowska E, Stepniak I, Krysa W, Ziora-Jakutowicz K, Rakowicz M, Sobanska A, et al. Next-generation sequencing study reveals the broader variant spectrum of hereditary spastic paraplegia and related phenotypes. Neurogenetics 2019;20:27–38.
- Nam DE, Yoo DH, Choi SS, Choi BO, Chung KW. Wide phenotypic spectrum in axonal Charcot–Marie–Tooth neuropathy type 2 patients with KIF5A mutations. Genes Genomics. 2018;40:77–84.
- He J, Liu X, Tang L, Zhao C, He J, Fan D. Whole-exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and Charcot-Marie-Tooth type 2. J Neurol Neurosurg Psychiatry. 2020;91:326–8.
- De Fuenmayor Fernández de La Hoz CP, Hernández-Laín A, Olivé M, Sánchez-Calvín MT, Gonzalo-Martínez JF, Domínguez-González C. Adult-onset distal spinal muscular atrophy: a new phenotype associated with KIF5A mutations. Brain. 2019;142:e66–e66.
- Brenner D, Yilmaz R, Müller K, Grehl T, Petri S, Meyer T, German ALS network MND-NET, et al. Hot-spot KIF5A mutations cause familial ALS. Brain. 2018;141:688–97.
- Zhang K, Liu Q, Shen D, Tai H, Liu S, Wang Z, et al. Mutation analysis of KIF5A in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging. 2019;73:229.e1-229–e4.
- Gu X, Li CYu, Chen Y, Wei Q, Cao B, Ou RWei, et al. Mutation screening of the KIF5A gene in Chinese patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90:245–6.
- Faruq M, Kumar D, Wadhwa S, Shamim U, Mathur A, Parveen S, et al. Intrafamilial variable spastic paraplegia/ataxia/ALS phenotype linked to a novel KIF5A mutation. Clin Genet. 2019;96:271–3.
- Simone M, Trabacca A, Panzeri E, Losito L, Citterio A, Bassi MT. KIF5A and ALS2 variants in a family with hereditary spastic paraplegia and amyotrophic lateral sclerosis. Front Neurol. 2018;9:1078.
- Tripolszki K, Gampawar P, Schmidt H, Nagy ZF, Nagy D, Klivényi P, et al. Comprehensive genetic analysis of a Hungarian amyotrophic lateral sclerosis cohort. Front Genet. 2019;10:732.
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–76.
- Lo Giudice M, Neri M, Falco M, Sturnio M, Calzolari E, Di Benedetto D, et al. A missense mutation in the coiled-coil domain of the KIF5A gene and late-onset hereditary spastic paraplegia. Arch Neurol. 2006;63:284–7.
- Jia X, Madireddy L, Caillier S, Santaniello A, Esposito F, Comi G, et al. Genome sequencing uncovers phenocopies in primary progressive multiple sclerosis. Ann Neurol. 2018;84:51–63.
- Guinto CO, Diarra S, Diallo S, Cissé L, Coulibaly T, Diallo SH, et al. A novel mutation in KIF5A in a Malian family with spastic paraplegia and sensory loss. Ann Clin Transl Neurol. 2017;4:272–5.
- Daud D, Griffin H, Douroudis K, Kleinle S, Eglon G, Pyle A, et al. Whole exome sequencing and the clinician: we need clinical skills and functional validation in variant filtering. J Neurol. 2015;262:1673–7.
- Lynch DS, Koutsis G, Tucci A, Panas M, Baklou M, Breza M, et al. Hereditary spastic paraplegia in Greece: characterisation of a previously unexplored population using next-generation sequencing. Eur J Hum Genet. 2016;24:857–63.