
Abstract
Objective: To evaluate the possible effect of reldesemtiv, a fast skeletal muscle troponin activator, on prescription and acceptance of durable medical equipment (DME) in the FORTITUDE-ALS trial. Methods: Health economic outcome information was collected in FORTITUDE-ALS (NCT03160898); sites recorded if and when DME, specifically manual or power wheelchairs, gastrostomy tubes, noninvasive ventilators, or augmentative language devices, was prescribed by a physician and accepted by the patient (DME-PAP) during the trial. Acceptance was defined as the patient agreeing the item was needed. Cox regression analysis compared time to DME-PAP for each reldesemtiv dose with placebo. Post hoc analyses evaluated all reldesemtiv doses compared with placebo. Results: At least one DME item was prescribed and accepted by 33/114 (28.9%) of placebo patients, 19/112 (17.0%) of patients receiving reldesemtiv 150 mg bid, 24/113 (21.2%) receiving 300 mg bid, and 29/117 (24.8%) receiving 450 mg bid. The proportion of new DME-PAP was significantly lower in patients receiving reldesemtiv 150 mg bid vs placebo (17.0% vs 28.9%, p = 0.032). The hazard ratio versus placebo for accepting at least one DME item for all reldesemtiv doses combined was 0.61 (confidence interval: 0.39, 0.96, p = 0.032). 25% of placebo patients were prescribed and agreed to obtain a DME item by 84 days; this threshold was met for reldesemtiv-treated patients at 120 days. Conclusions: Results suggest ALS patients receiving reldesemtiv may have lower risk of and delayed need for DME related to impaired mobility, breathing, swallowing, or speaking; this delay is consistent with other measures indicating delay in disease progression.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease associated with significant social and economic burden on both the patients and their caregivers. Without a cure, treatment is primarily supportive, and medical and assistive needs invariably increase over the course of the disease progression (Citation1,Citation2). Effective therapies for patients with ALS may preserve quality of life and reduce costs for these patients. These potential benefits of treatment may also delay the time to reach disease-related functional milestones, such as requiring durable medical equipment (DME). Previous reports suggest that 46–96% of patients with ALS will require a variety of DME depending on the site of onset, with 55% of patients with ALS requiring five or more assistive technology devices (Citation2,Citation3). Delaying the time to need items of DME would permit the patient to be more independent for a longer period of time.
Reldesemtiv is a second-generation fast skeletal muscle troponin activator (FSTA) that sensitizes the sarcomere to calcium leading to increased muscle force, particularly at low- to mid-range stimulation frequencies, intended to improve physical functions such as walking, breathing, and swallowing. A single-dose study in healthy participants showed that reldesemtiv had a greater pharmacodynamic effect on muscle force generation with submaximal nerve stimulation frequencies than was observed in an earlier study with tirasemtiv (a first-generation FSTA) without central nervous system side effects (Citation4,Citation5). This led to the design of FORTITUDE-ALS (Functional Outcomes in a Randomized Trial of Investigational Treatment with CK-2127107 to Understand Decline in Endpoints in ALS; NCT03160898), a phase 2b clinical trial evaluating the safety, tolerability, and efficacy of three doses of reldesemtiv compared with placebo in patients with ALS (Citation6). This randomized, double-blind, placebo-controlled clinical trial did not demonstrate a statistically significant difference between reldesemtiv and placebo for the dose–response analyses of the primary endpoint (change from baseline in slow vital capacity [SVC] at 12 weeks), key secondary endpoints (change from baseline in the ALS Functional Rating Scale-Revised [ALSFRS-R] and slope of the muscle mega-score from baseline to 12 weeks). However, a pre-specified analysis of the two highest doses of reldesemtiv combined, and a post hoc analysis of all doses of reldesemtiv combined compared with placebo, revealed statistically significant differences favoring reldesemtiv in the change from baseline to Week 12 of the ALSFRS-R Total Score.
We wished to evaluate the potential health economic impact of reldesemtiv by investigating the time to prescription and acceptance of DME items and the number of patients who were prescribed and agreed to obtain at least one DME item while participating in FORTITUDE-ALS. The DME items specifically recorded were manual wheelchair, power wheelchair, gastrostomy tube, noninvasive ventilator (NIV), or augmentative language device. The endpoint was the time to have the DME prescribed by the physician and accepted by the patient as needed (DME-PAP). This endpoint was selected (rather than the actual receipt of, or first use of the DME) given the relatively short trial duration and the variability across insurance coverages and countries in the time to receive the DME once prescribed.
Materials and methods
Trial design
FORTITUDE-ALS (NCT03160898) was a phase 2b, randomized, double-blind, placebo-controlled, dose-ranging trial conducted at 65 sites in the United States (n = 283), Canada (n = 100), Ireland (n = 4), Spain (n = 38), the Netherlands (n = 11), and Australia (n = 20) (Citation6). The trial evaluated the safety, tolerability, and efficacy of three doses of reldesemtiv compared with placebo in patients with ALS (Citation6). All sites received institutional review board approvals before enrollment, and all patients provided written informed consent. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
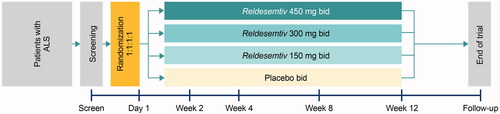
The methodology of FORTITUDE-ALS has been reported (Citation6). Briefly, patients were eligible if they were between 18 and 80 years of age and had a diagnosis of ALS established within the past 24 months. Patients were required to have an upright SVC ≥ 60% predicted for age, height, sex, and ethnicity at screening, and to be either not taking or on stable doses of riluzole (for ≥ 30 days before screening) and/or edaravone (completed two or more cycles before screening). Patients were excluded from the trial if at screening they used NIV, could not swallow a whole tablet, had prior use of reldesemtiv or tirasemtiv, or had received stem cell or gene therapy for ALS. The trial had an active treatment period of 12 weeks and a 4-week follow-up period after the last dose of double-blind study drug. Patients (N = 457) were randomized (1:1:1:1) and treated with reldesemtiv 150, 300, or 450 mg twice daily (bid), or placebo ().
Figure 1. FORTITUDE-ALS trial design. ALS: amyotrophic lateral sclerosis; bid: twice daily.
DME-PAP assessment and post hoc analysis
Information on health economic outcomes was collected for all patients in FORTITUDE-ALS, recording if and when at least one of the following pieces of DME was prescribed and accepted by the patient during the course of the trial: manual wheelchair, power wheelchair, gastrostomy tube, noninvasive ventilator, or augmentative communication device. Augmentative communication devices were not pre-defined; therefore, they may have included devices ranging from an off-the-shelf tablet with text-to-voice capability to a sophisticated built-for-purpose speech-generating device activated through eye gaze. A pre-specified Cox regression analysis was used to compare time to first prescription and acceptance of any new DME in each reldesemtiv dose group with placebo, stratifying for riluzole or edaravone use in the full analysis set (FAS). The FAS consisted of all randomized patients who received any study drug and had a baseline and at least one post-baseline efficacy assessment during the double-blind period. Post hoc analyses compared the use of DME for all reldesemtiv doses combined compared with placebo. In the Cox regression analysis, treatment groups were fixed terms with baseline scores of ALSFRS-R items 1, 3, 5, and 8 as covariates, stratifying for the following factors: pooled sites, riluzole use at baseline, and edaravone use at baseline. If a patient terminated early and without the event of DME-PAP, the patient was censored at the date of last contact. If a patient terminated early and had DME-PAP before early termination, the patient was counted as having DME-PAP at the time of DME-PAP before early termination. p values were generated from a Chi-square test.
Results
Patient disposition and baseline characteristics
As reported in Shefner et al. (Citation6), the mean age of patients was 58.7 years, the mean number of months since diagnosis was 8.6 months, and 60.5% of patients were male. The overall mean ALSFRS-R Total Score was 37.4, and mean SVC was 84.7% of predicted (Citation6). The site of onset of ALS was similar across patients treated with placebo compared with all reldesemtiv doses combined (upper limb onset: 42.1%, 44.4%; lower limb onset: 38.6%, 36.5%; bulbar onset: 19.3%, 19.0%, respectively).
In order to determine if there was imbalance between the placebo and reldesemtiv groups regarding their use of specific DME items at baseline, we reviewed responses to ALSFRS-R item 1 (speech), item 3 (swallowing), item 5B (used when a patient has a feeding tube), and item 8 (walking) (). Patients who scored 0 or 1 on item 1 may have sufficiently impaired speech to necessitate an augmentative language device; these scores were recorded in similar percentages of patients on reldesemtiv (5.6%) and placebo (4.4%). A score of 0 or 1 on item 3 would indicate the patient had a gastrostomy tube, as would using item 5B instead of item 5A to answer questions about using eating utensils. Only one patient, who was in the reldesemtiv group, had a gastrostomy tube. Patients who were non-ambulatory and therefore required a wheelchair would score 0 or 1 on item 8. Being non-ambulatory was more common in patients on placebo (13.2%) than on reldesemtiv (5.3%).
Table 1. Baseline ALSFRS-R scores for items 1, 3, 5, and 8 for patients in the placebo and reldesemtiv groups.
Overall, 105/456 (23.0%) patients were prescribed and accepted at least one DME; of these, 33/114 (28.9%) patients were randomized to placebo, 19/112 (17.0%) patients were receiving reldesemtiv 150 mg bid, 24/113 (21.2%) were receiving 300 mg bid, and 29/117 (24.8%) were receiving 450 mg bid.
DME-PAP
The probability of new DME-PAP was delayed for all doses of reldesemtiv compared with placebo, with the highest probability of no new DME over time for patients randomized to reldesemtiv 150 mg bid (). For all reldesemtiv doses combined, the probability of new DME-PAP was significantly reduced compared with placebo (p = 0.032; ). Twenty-five percent of patients given placebo had been prescribed and accepted a DME item by 84 days while 25% of patients treated with reldesemtiv had not been prescribed and accepted a DME item until 120 days. The calculated value of 120 days for all reldesemtiv doses combined was longer than the planned 112-day duration of the study (i.e. from the first dose of double-blind study drug to the end of the follow-up period), because the final follow-up study visits were sometimes scheduled more than 4 weeks after the last dose of double-blind study drug.
Figure 2. Probability of no new DME-PAP over time with reldesemtiv treatment compared with placebo. (A) Each dose of reldesemtiv, and (B) for all doses of reldesemtiv combined. Numbers of patients per group are indicated on top of the x-axis. bid: twice daily; DME-PAP: durable medical equipment prescribed and accepted by the patient.
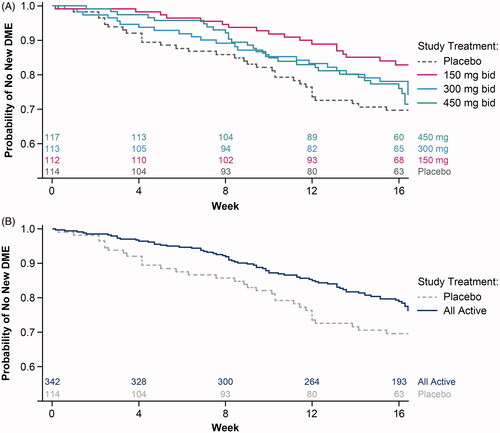
Compared with placebo, the hazard ratio (HR) for the time to first DME-PAP was significantly reduced for patients randomized to reldesemtiv 150 mg bid (HR = 0.43 [95% confidence interval: 0.23, 0.79, p = 0.006]) (Citation6), and was numerically reduced for patients randomized to reldesemtiv 300 mg bid and 450 mg bid (). The HR for the time to first DME-PAP was significantly lower for all reldesemtiv doses combined (HR = 0.61 [CI: 0.39, 0.96], p = 0.032) compared with placebo. Thus, for patients taking reldesemtiv, there was a 39% lower chance of being prescribed and accepting new DME over the course of the trial.
Table 2. Hazard ratiosa for reldesemtiv compared with placebo for at least one DME-PAP over the course of the trial.
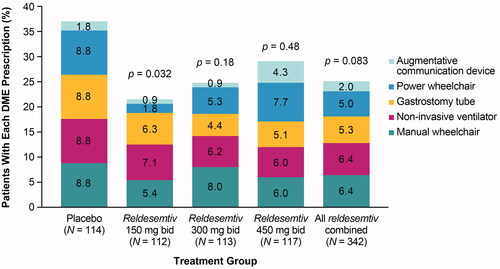
The proportion of new DME-PAP was significantly lower in the reldesemtiv 150 mg bid group (19/112 [17.0%], p = 0.032) and was numerically reduced in patients randomized to reldesemtiv 300 mg bid (24/113 [21.2%], p = 0.18), reldesemtiv 450 mg bid (29/117 [24.8%], p = 0.48), and all reldesemtiv-treated groups combined (72/342 [21.1%], p = 0.083) compared with placebo (33/114 [28.9%]; , ). No one type of DME appeared to drive this result; with the exception of the augmentative communication device (which was prescribed and agreed to by the smallest numbers of patients for all doses), the percentage of each new DME for patients receiving placebo was numerically higher than for all reldesemtiv doses combined.
Figure 3. Proportion of DME-PAP by treatment group over the course of the trial. bid: twice daily; DME-PAP: durable medical equipment prescribed and accepted by the patient. p Values for reldesemtiv versus placebo were obtained from a Chi-square test.
Discussion
Progression of ALS is often accompanied by difficulties with ambulation, breathing, speech, and swallowing. DME such as wheelchairs, noninvasive ventilators, augmentative communication devices, and gastrostomy tubes are frequently required as the disease progresses, and dependency on these items may be viewed as a disability milestone. Therapies that slow disease progression may delay the burden of DME for patients. It would be logical to presume that treatments that can improve patients’ level of physical function and extend the time to the need for DME would at least maintain, if not improve, the quality of life in patients with ALS. In this post hoc analysis of data from a randomized double-blind trial, compared with placebo, the probability of new DME-PAP was significantly reduced for all doses of reldesemtiv combined, and reldesemtiv treatment delayed the 25th percentile time to patients being prescribed and accepting DME by 36 days. Patients receiving reldesemtiv had a significantly (39%) lower chance of being prescribed and accepting DME related to impaired mobility, breathing, swallowing, or speaking compared with placebo. The results of this post hoc analysis indicate that there is a longer time to DME being prescribed and accepted by patients receiving reldesemtiv relative to placebo.
We believe this is the first ALS clinical trial to analyze the time to have a physician prescribe and the patient agree to use an item of DME. We chose to utilize this novel outcome measure given the rising emphasis on health economics outcome research as well as the focus on outcome measurements that are clinically meaningful to the patient. For patients, delaying the time to requiring wheelchairs, augmentative communication devices, feeding tubes, or noninvasive ventilation are endpoints that are easily understood since independence and the resultant impact on caregiver burden are issues with which all patients with ALS struggle.
We recognize that there are multiple factors that influence procurement and use of DME. It is well known that the timing of NIV is quite variable both by country and by treating physician, and healthcare systems vary regarding how easy or difficult it may be to qualify for insurance coverage for NIV. Previous reports show that about half of patients with ALS experience difficulties procuring DME, most often because of long delivery times and waiting lists (Citation7). There is a high failure rate in the procurement of assistive technology devices crucial for the management of ALS, including powered wheelchairs (52%) and communication devices (39%). Some of the most common causes for this failed procurement of essential equipment include having coverage declined by health insurance providers (50.9%), the DME refused by the patient (29.5%), or death of the patient before equipment delivery (19.6%) (Citation3). Patient reasons for refusing DME include worries about increased reliance on caregivers; DME use may also reinforce perceptions of deterioration in their own physical condition (Citation2,Citation3,Citation8). In addition, the willingness of patients to accept the need for DME may also be influenced by out-of-pocket expenses and personal circumstances (such as the availability of a van to transport a power wheelchair). However, there is no reason to expect that these issues would differentially impact the patients in the various treatment arms.
This analysis has a few limitations. Given the short trial duration, a limited number of patients were prescribed and agreed to obtain DME items. This short trial duration may also have contributed to the non-significant but numerical differences between the placebo and reldesemtiv 300 mg bid and 450 mg bid groups; perhaps a longer follow-up would have revealed significant differences over time for all doses. The small portion of patients with DME data also made it difficult to explore possible trends related to the site of ALS onset. Another limitation in interpretation of these data is that baseline DME items was not definitively established. Patients were not using NIV at baseline, as per inclusion/exclusion criteria, but whether patients already had a gastrostomy tube but were still able to swallow a tablet whole, or had a wheelchair or augmentative communication device (which also could be highly heterogeneous) was not recorded. Comparison of the groups showed that similar percentages of patients in the reldesemtiv and placebo groups were likely to be using an augmentative communication device (i.e. scored 0 or 1 on item 1), and that one patient had a gastrostomy tube at baseline. We could not distinguish between manual or power wheelchair use at baseline, nor could we distinguish patients who had a wheelchair but were still ambulatory. However, being non-ambulatory was about 8% more common in patients on placebo than those on reldesemtiv, meaning that there were more patients on reldesemtiv at risk for losing ambulation. In order to address the possible imbalance in DME use at baseline between the placebo and reldesemtiv groups, we included baseline ALSFRS-R items 1, 3, 5, 8 as covariates in the Cox model. This technique reasonably estimates baseline DME use for augmentative communication devices and wheelchairs and is a direct way to determine baseline gastrostomy use without the basis of randomization.
Our data suggest that DME prescription and use may be a useful novel outcome measure in ALS trials. Future research may include collecting data for when different DME items are first prescribed, obtained, and used, as this may highlight the challenges and delays experienced by patients with ALS obtaining DME. Longer trials would also permit collection of how extent of use may change over time, and further investigate onset site as it relates to which DME items are used early compared to later in the disease course. Recording reasons for obtaining DME may also help to better understand the variability that may be seen in DME utilization. Given that there is a wide range of different types of augmentative and assistive communication devices that may be used by a patient with a similarly broad range of cost, collecting more detailed information on the specific device would be useful for a cost analysis. Additional studies are also warranted to investigate the cost–benefit relationship of a reduced risk of DME use, and the effect of delayed requirement for DME on the quality of life for patients with ALS.
The authors acknowledge Ciara Duffy, PhD (Evidence Scientific Solutions, Horsham, UK) and Richard Fay, PhD, CMPP (Evidence Scientific Solutions, Philadelphia, PA, USA) for writing assistance which was funded by Cytokinetics, Incorporated.
The FORTITUDE-ALS study group
Jinsy A. Andrews, MD, The Eleanor and Lou Gehrig ALS Center, The Neurological Institute, Columbia University, New York, NY; Bettina M. Cockroft, MD, Cytokinetics, Incorporated, South San Francisco, CA; Cynthia Bodkin, MD, FAAN, Indiana University Health, Indianapolis, IN; Benjamin Brooks, MD, Atrium Health Neurosciences Institute - Carolinas Neuromuscular/ALS Center, Charlotte, NC; James Caress, MD, Wake Forest Health Sciences, Winston-Salem, NC; Annie Dionne, MD, FRCPC, CHU de Québec-Université Laval, Quebec, QC; Dominic Fee, MD, FAAN, Medical College of Wisconsin, Milwaukee, WI; Angela Genge, MD, Montreal Neurological Institute, Montreal, QC; Stephen A. Goutman, MD, MS, University of Michigan, Ann Arbor, MI; Namita A. Goyal, MD, UC Irvine ALS & Neuromuscular Center, Orange, CA; Orla Hardiman, MD, Trinity College, Beaumont Hospital, Dublin, Ireland; Ghazala Hayat, MD, FAAN, FANA, St. Louis University, St. Louis, MO; Terry Heiman-Patterson, MD, Lewis Katz School of Medicine, Temple University, Philadelphia, PA; Daragh Heitzman, MD, FAAN, Texas Neurology, Dallas, TX; Robert D. Henderson, MBBS (Hons), PhD, Royal Brisbane and Women's Hospital, Herston, Australia; Carlayne Jackson, MD, University of Texas Health Science Center, San Antonio, TX; Wendy Johnston, MD, FRCPC, University of Alberta, Edmonton, AB; Chafic Karam, MD, Oregon Health & Science University, Portland, OR; Matthew C. Kiernan, MBBS (Hons), PhD, DSc, FRACP, FAHMS, University of Sydney, Royal Prince Alfred Hospital, Sydney, Australia; Stephen J. Kolb, MD, PhD, Ohio State University Wexner Medical Center, Columbus, OH; Lawrence Korngut, MD, MSc, FRCPC, University of Calgary, Calgary, AB; Shafeeq Ladha, MD, St. Joseph's Hospital and Medical Center, Barrow Neurological Institute, Phoenix, AZ; Noah Lechtzin, MD, Johns Hopkins School of Medicine, Baltimore, MD; Fady I. Malik, MD, PhD, Cytokinetics, Incorporated, South San Francisco, CA; Genevieve Matte, MDCM, FRCPC, Center Hospitalier de l’Université de Montréal, Montreal, QC; Lisa Meng, PhD, Cytokinetics, Incorporated, South San Francisco, CA; Timothy M. Miller, MD, PhD, Washington University School of Medicine, St. Louis, MO; Jesus S. Mora, MD, Hospital San Rafael, Madrid, Spain; Merrilee Needham, MBBS, PhD, FRACP, Perron Institute, Department of Neurology Fiona Stanley Hospital, The University of Notre Dame Australia, Murdoch University, Perth, Australia; Bjorn Oskarsson, MD, Mayo Clinic Florida, Jacksonville, FL; Gary Pattee, MD, Neurology Associates, Lincoln, NE; Erik P. Pioro, MD, PhD, FRCPC, Cleveland Clinic, Cleveland, OH; Michael Pulley, MD, PhD, University of Florida, Jacksonville, FL; Dianna Quan, MD, University of Colorado Denver, Aurora, CO; Kourosh Rezania, MD, University of Chicago Medical Center, Chicago, IL; Stacy A. Rudnicki, MD, Cytokinetics, Incorporated, South San Francisco, CA; Jeremy M. Shefner, MD, PhD, Barrow Neurological Institute, Phoenix, AZ; Kerri Schellenberg, MD, University of Saskatchewan, Saskatoon, SK; David Schultz, BMBS, Flinders Medical Center, Bedford Park, Australia; Christen Shoesmith, MD, FRCPC, London Health Sciences Center, London, ON; Zachary Simmons, MD, Penn State Hershey Medical Center, Hershey, PA; Jeffrey Statland, MD, University of Kansas Medical Center, Kansas City, KS; Shumaila Sultan, MD, West Virginia University, Morgantown, WV; Andrea Swenson, MD, University of Iowa City, IA; Leonard H. van den Berg, MD, PhD, Department of Neurology, UMC Utrecht Brain Center, University Medical Center Utrecht, Utrecht, The Netherlands; Tuan Vu, MD, University of South Florida, Tampa, FL; Steve Vucic, MBBS (Hons I), PhD, DSc, FRACP, Westmead Hospital, Sydney, Australia; Jenny Wei, PhD, Cytokinetics, Incorporated, South San Francisco, CA; Michael Weiss, MD, University of Washington, Seattle, WA; Ashley Whyte-Rayson, MD, Duke University School of Medicine, Durham, NC; Andrew A. Wolff, MD, Cytokinetics, Incorporated, South San Francisco, CA; James Wymer, MD, PhD, University of Florida, Gainesville, FL; Lorne Zinman, MD, MSc, FRCPC, University of Toronto, ALS/Neuromuscular Clinic Sunnybrook Health Sciences Center, Toronto, ON.
Supplemental Material
Download MS Word (24.5 KB)Acknowledgements
We wish to thank the participants of FORTITUDE-ALS and their families for their contributions to this clinical trial, the investigators of FORTITUDE-ALS, and members of the Data Monitoring Committee and Steering Committee.
Declaration of interest
JAA has served as consultant for AL-S Pharma, Avexis, Biogen, Cytokinetics, Denali and Wave Live Sciences; has received research support from Biogen, Novartis, Orion and NIH; and is a former employee of Cytokinetics.
AG has served as a consultant for AB Sciences, AL-S Pharma, Avexis, Biogen, Cytokinetics, Incorporated, MT Pharma America, and Roche.
CJ has served as a consultant for Argenex, Cytokinetics, Incorporated, ITF Pharma, MT Pharma America, and Strongbridge Pharmaceuticals; served on Speaker’s Bureau for Avanir, CSL Behring, MT Pharma America, and Strongbridge Pharmaceuticals; has received research support from Amylyx, Cytokinetics, Incorporated, and NIH; and is currently serving as a member of a Data Safety Monitoring Board for Anelixis, Brainstorm, and Mallinckrodt.
NL has served as a consultant/advisor for Cytokinetics, Incorporated, Hill-Rom, and Vertex; and has received research support from AstraZeneca and Vertex.
TMM has served as a consultant/advisor for Biogen and Cytokinetics, Incorporated; has received research support from Biogen and Ionis; and receives licensing fees from C2N.
JMS has served as a consultant for Biogen, Biohaven, Cytokinetics, Incorporated, MT Pharma America, and Novartis; has received research support from Amylyx, Biogen, Biohaven, Biotie, Cytokinetics, Incorporated, MT Pharma America, Neuraltus, and Orphazyme; and has received compensation from UpToDate for serving as neuromuscular section editor.
BMC was an employee of Cytokinetics, Incorporated at the time of the study and owns stock in Cytokinetics, Incorporated. SAR, FIM, LM, JW, and AAW are employees of and own stock in Cytokinetics, Incorporated.
Data availability statement
Data reported herein are part of a sponsor-led clinical development program that is ongoing, and thus complete datasets for the trial will not be made available with this report.
Additional information
Funding
Related Research Data
References
- Gladman M, Zinman L. The economic impact of amyotrophic lateral sclerosis: a systematic review. Expert Rev Pharmacoecon Outcomes Res. 2015;15:439–50.
- Bromberg MB, Brownell AA, Forshew DA, Swenson M. A timeline for predicting durable medical equipment needs and interventions for amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler. 2010;11:110–15.
- Funke A, Spittel S, Grehl T, Grosskreutz J, Kettemann D, Petri S, et al. Provision of assistive technology devices among people with ALS in Germany: a platform-case management approach. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:342–50.
- Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, et al. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018;57:729–34.
- Hansen R, Saikali KG, Chou W, Russell AJ, Chen MM, Vijayakumar V, et al. Tirasemtiv amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2014;50:925–31.
- Shefner JM, Andrews JA, Genge A, Jackson C, Lechtzin N, Miller TM, et al. A phase 2, double-blind, randomized, dose-ranging trial of reldesemtiv in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:287–99.
- Creemers H, Beelen A, Grupstra H, Nollet F, van den Berg LH. The provision of assistive devices and home adaptations to patients with ALS in the Netherlands: patients' perspectives. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:420–25.
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205–15.