
Abstract
Objective:To determine if inflammation in proximity of the motor unit may contribute to neurodegeneration in amyotrophic lateral sclerosis (ALS). Methods: We identified all patients diagnosed in Sweden with concurrent ALS and multiple sclerosis (MS), myasthenia gravis (MG), inflammatory polyneuropathies (IP), or dermatopolymyositis (DMPM) during 1991–2014 according to the Swedish Patient Register (N = 263). We validated medical records for 92% of these patients (18 records were not retrieved and three did not contain enough information) and compared patients with a confirmed overlap (N = 28) with an independent sample of patients with solely ALS (N = 271). Results: Ninety-one patients were deemed as not having ALS (34.6%). Among the remaining 151 with validated ALS, 12 had also a confirmed MS diagnosis, nine a confirmed MG diagnosis, four a confirmed IP diagnosis, and three a confirmed DMPM diagnosis. Seventeen of the patients were women and 11 were men. Seventy-nine percent of the patients with a confirmed overlap had MS, MG, IP, or DMPM diagnosed prior to ALS. Compared to patients with only ALS, the concurrent patients were significantly older at symptoms onset, had higher prevalence of bulbar onset, but used Riluzole and noninvasive ventilation less frequently. Conclusions: We found that a high concurrence of ALS and MS/MG/IP/DMPM diagnoses is largely due to diagnostic uncertainty. A minority of patients had a true concurrence, where MS, MG, IP, and DMPM preceded the ALS diagnosis, which might be due to chance alone. Four patients were diagnosed with MG shortly after onset of ALS, suggesting that neurodegeneration might trigger autoimmunity.
Introduction
Although early symptoms of amyotrophic lateral sclerosis (ALS) may mimic multiple sclerosis (MS), myasthenia gravis (MG), inflammatory polyneuropathies (IP), or dermatopolymyositis (DMPM) (Citation1), biological overlap among these diseases may also exist (Citation2,Citation3). Such biological overlap may be due to shared etiologies, but also a causal relationship between the inflammation around the motor unit associated with MS/MG/IP/DMPM and neurodegeneration.
A study from the United Kingdom investigated five concurrent ALS/MS patients and proposed that MS-associated neuroinflammation could affect penetrance of the C9ORF72 expansion (Citation2). Larger epidemiological studies have confirmed the association between MS and ALS (Citation3,Citation4) and found an increased risk for MS among children of ALS patients (Citation5,Citation6).
The concurrence between ALS and MG has also been reported (Citation3,Citation7–14). The cohort study that examined the risk of ALS in people with MS, found that MG and polymyositis were also associated with later ALS risk (Citation4). The association of dermatomyositis and polymyositis with ALS was later confirmed in other populations (Citation3,Citation15). In contrast, the overlap between ALS and IP such as Guillain-Barré syndrome (GBS) has been reported only in the Swedish population (Citation3).
With an extensive nationwide medical records review we aimed to determine to what extent concurrence of an ALS diagnosis together with MS/MG/IP/DMPM is due merely to diagnostic uncertainty. To investigate the hypothesis of a biological overlap between these neuroinflammatory disorders and ALS, we also examined the temporal relationship between the diagnoses among patients with a confirmed overlap and compared their clinical characteristics to patients with only ALS.
Methods
Data collection and validation
The Swedish National Board of Health and Welfare collects in the Swedish Patient Register (Citation16) information about all hospital diagnoses given in Sweden (inpatient care since 1964 and outpatient care since 2001). From the Swedish National Board of Health and Welfare we obtained a complete list of personal identity numbers (Citation17) of all patients diagnosed in Sweden with concurrent ALS and MS/MG/IP/DMPM according to the 9th and 10th Swedish revisions of the International Classification of Diseases codes during 1991–2014 (N = 263; ). We selected the list of chronic inflammatory diseases of the motor unit to test our hypothesis that there is a biological overlap between neuroinflammation in close proximity to the motor unit and motor neuron degeneration in ALS. We sent the list of personal identity numbers to all the hospitals which the patients visited and requested to receive a copy of their medical records. Three neurologists specialized in ALS and neuroinflammation reviewed the medical records independently and made diagnostic accuracy decisions by consensus. ALS diagnosis was validated according to the revised El-Escorial criteria (Citation18). We included primary lateral sclerosis (PLS) in our definition of ALS given the increasing recognition as a sub-phenotype of ALS (Citation19). MS diagnosis was validated according to the 2017 revised McDonald criteria (Citation20) and the aid of imaging scans such as magnetic resonance imaging and analysis of cerebrospinal fluid. Because acetylcholine receptor antibodies (AchR) are not 100% specific for MG (Citation21,Citation22), the validation of MG diagnosis was made after taking into account the presence of AchR or muscle-specific tyrosine kinase antibodies (MuSK), neurophysiological signs of neuromuscular transmission defect, and a positive response to cholinesterase inhibitors. The IP diagnoses were validated according to the 2010 European Federation of Neurological Societies criteria for Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) (Citation23) and Multifocal Motor Neuropathy (MMN) (Citation24). In addition to the clinical picture, a GBS diagnosis was validated with the aid of cerebrospinal fluid analysis and nerve conduction studies (Citation25). Validation of a DMPM diagnosis was mainly based on histopathological verification of muscle biopsies.
Table 1 International Classification of Diseases (ICD) codes used to identify amyotrophic lateral sclerosis, multiple sclerosis, myasthenia gravis, inflammatory polyneuropathies, and dermatopolymyositis in the Swedish Patient Register.
Data analysis
We summarized clinical characteristics of the patients with a validated concurrent ALS and MS/MG/IP/DMPM diagnosis, including the temporal pattern of the diagnoses. To determine if patients with concurrent ALS and MS/MG/IP/DMPM had specific clinical characteristics of ALS, we compared them to an independent sample of patients diagnosed with only ALS (N = 271) selected from the Swedish Patient Register, who had visited a hospital concerning ALS during 2013–2014 in Stockholm. A diagnostic validation and detailed medical record review of these patients had already been performed (Citation26). We tested differences between the two groups by Chi-square test (categorical variables), Fisher exact test (categorical variables with expected frequencies ≤5), or Wilcoxon test (non-normally distributed continuous variables). We also conducted a secondary analysis after excluding PLS patients from our definition of ALS.
We considered statistically significant differences with a 2-sided P-value of ≤0.05 and performed analyses using Stata software, version 15.1 (StataCorp, College Station, TX).
Participants of this study did not agree for their data to be shared publicly, so supporting data are not available. The Regional Ethical Review Board in Stockholm, Sweden, granted ethical permit for this project and waived us from the need of seeking informed consent, given that the majority of study participants were no longer alive at the time of data collection.
Results
Data collection and validation
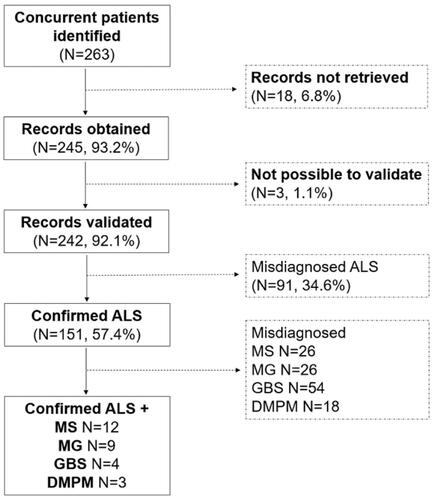
The hospitals were unable to retrieve medical records for 18 patients (6.8%). Hence, we obtained medical records of 245 patients with a registered diagnosis of concurrent ALS and MS/MG/IP/DMPM (93.2%). Three of the medical records retrieved did not contain enough information to make a decision on diagnostic accuracy (1.1%).
Ninety-one patients were deemed as not having ALS (34.6%), but dementia (N = 1), inclusion body myositis (N = 16), inflammatory polyneuropathy (N = 21), Kennedy's disease (N = 3), MG (N = 8), MG and polyneuropathy (N = 1), MG and spinal stenosis (N = 1), mononeuritis (N = 1), MS (N = 15), MMN (N = 4), myelitis (N = 1), myelopathy (N = 2), myositis (N = 3), Parkinson’s disease (N = 1), peroneal nerve paralysis (N = 1), polyneuropathy (N = 8), spinal stenosis (N = 2), or spinocerebellar ataxia (N = 2).
Among the remaining 151 patients with confirmed ALS, we additionally confirmed a MS diagnosis in 12, MG in nine, IP in four, and DMPM in three patients (: flow-chart of data collection and diagnostic accuracy decisions).
Figure 1 Flow-chart of data collection and diagnostic accuracy decisions.
Clinical characteristics of patients with confirmed concurrent ALS and MS/MG/IP/DMPM diagnosis
We report the clinical characteristics of each patient diagnosed with confirmed concurrent ALS and MS/MG/IP/DMPM (N = 17 women and 11 men; median age at ALS diagnosis 66.5 years, range 24–86; ). Patients with confirmed concurrent ALS and MS/MG/IP/DMPM were diagnosed with ALS during 1997–2013.
Table 2 Clinical characteristics of the patients diagnosed with concurrent amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS), myasthenia gravis (MG), inflammatory polyneuropathies (IP), or dermatopolymyositis (DMPM).
Three of these patients were diagnosed with PLS and MS, and two were diagnosed with PLS and MG.
Seventy-nine percent of the patients with a confirmed overlap had MS/MG/IP/DMPM diagnosed prior to ALS (N = 22, median time 6 years, range 0–53). Only a minority of patients (N = 6, 21%) were diagnosed first with ALS and then with MS (N = 2) or MG (N = 4), after a median interval < one year (range 0–8).
We assessed subtypes of MS among nine of the 12 patients with confirmed ALS/MS overlap (66.7%). Three of them had Primary-Progressive MS (PPMS), three had Secondary-Progressive MS (SPMS), and three had Relapsing-Remitting MS (RRMS). Seven of the nine patients with a confirmed ALS/MG overlap had generalized MG (77.8%) and two had bulbar MG (22.2%). Eight of the nine ALS/MG patients were tested for antibodies against AchR, of which 75% (N = 6) were positive. No patient had MuSK antibodies.
Additionally, five patients with a confirmed concurrent ALS/MG showed pathological decrement upon repetitive nerve stimulation (five out of seven patients with reported neurophysiological test results, 71.4%). We assessed subtypes of IP for three of the four ALS/IP patients identified, two had the GBS variant Acute Motor Axonal Neuropathy (AMAN; 66.7%) and one had CIDP (33.3%). Two of the three ALS/DMPM patients had polymyositis (66.7%) and one had unspecified myositis (33.3%).
Nine patients were still alive according to the latest medical records we received (32.1%), their median (range) disease duration from ALS diagnosis to the latest medical record was 6.7 (Citation1–13) years.
We were not able to report age at ALS onset and ALS diagnostic delay of patients with uncertain date of ALS onset (N = 14, 50%).
Comparison between patients with a confirmed concurrent ALS and MS/MG/IP/DMPM diagnosis and patients with only ALS
Compared to patients with only ALS, patients with concurrent ALS and MS/MG/IP/DMPM had a higher median age at ALS onset (70.5 vs. 62 years; p = 0.014), a higher prevalence of bulbar ALS onset (46.4% vs. 28.8%; p = 0.037), a lower prevalence of Riluzole use (64.3% vs. 87.8%; p = 0.001), but a shorter median time between ALS diagnosis and Riluzole prescription if used (1 month vs. 19 months; p < 0.0001), and a lower prevalence of noninvasive ventilation use (17.9% vs. 48.0%; p = 0.002; ).
Table 3 Patients diagnosed with concurrent amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS), myasthenia gravis (MG), inflammatory polyneuropathies (IP), or dermatopolymyositis (DMPM) significantly differ in terms of age at ALS symptoms onset, site of ALS onset, use of Riluzole, time from ALS diagnosis to first Riluzole prescription, and use of noninvasive ventilation as compared to patients diagnosed with only ALS.
Specifically, patients with concurrent ALS and MS had a lower prevalence of Riluzole use (58.3% vs. 87.8%; p = 0.013), shorter median time between ALS diagnosis and Riluzole prescription if used (<1 month vs. 19 months; p = 0.005), enteral nutrition use (8.3% vs. 46.9%; p = 0.014), and noninvasive ventilation use (0.0 vs. 48.0%; p = 0.001), compared to patients with only ALS. Patients with concurrent ALS and MG had a higher median age at ALS onset (73 years vs. 62 years; p = 0.015) and at diagnosis (75 years vs. 64 years; p = 0.024), a higher prevalence of bulbar ALS onset (77.8% vs. 28.8%; p = 0.001; ), and a shorter median time between ALS diagnosis and Riluzole prescription if used (<1 month vs. 19 months; p = 0.0002), compared to patients with only ALS. Patients with concurrent ALS and DMPM had also a shorter median time between ALS diagnosis and Riluzole prescription if used (<1 month vs. 19 months; p = 0.0002), compared to patients with only ALS.
There were no statistically significant differences between patients with concurrent ALS and MS/MG/IP/DMPM and patients with only ALS in terms of sex, ALS diagnostic delay, cognitive impairment before diagnosis, invasive ventilation use, or survival ().
Excluding PLS patients from our definition of ALS (N = 3 with PLS and MS, N = 2 with PLS and MG, and N = 17 from the comparison group with only ALS) did not significantly alter the differences between groups in the main analysis ().
Table 4 Patients diagnosed with concurrent amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS), myasthenia gravis (MG), inflammatory polyneuropathies (IP), or dermatopolymyositis (DMPM) significantly differ in terms of age at ALS symptoms onset, site of ALS onset, use of Riluzole, time from ALS diagnosis to first Riluzole prescription, and use of noninvasive ventilation as compared to patients diagnosed with only ALS. Primary lateral sclerosis was not included in our definition of ALS in this analysis.
Discussion
Using validated data from a nationwide medical records review during 1991–2014 in Sweden, we found that most of the overlap between ALS and MS/MG/IP/DMPM was due to misdiagnosis, while 28 patients were confirmed having concurrent ALS and MS/MG/IP/DMPM. These patients were older at ALS onset, had higher prevalence of bulbar onset, and were treated with Riluzole and noninvasive ventilation less frequently, compared to patients with only ALS. Most of these patients developed their neuroinflammatory disorders years before ALS onset.
Clinical characteristics of patients with confirmed concurrent ALS and MS/MG/IP/DMPM diagnosis and comparison with patients with only ALS
Prior studies have reported a concurrence between ALS and MS and ALS and MG, suggesting a link between these diseases (Citation2,Citation3,Citation7–14). We here also identified three patients with concurrent ALS and DMPM, and four with ALS and GBS and ALS and CIDP. While this may suggest shared etiological factors or that chronic or acute neuroinflammation around the motor unit increases susceptibility for ALS, it may also be due to chance alone. Moreover, we recently found no supporting evidence of shared genetic mechanisms between autoimmune diseases and ALS (Citation3), which argues against the suggested overlap to be caused by hereditability.
We made the unique effort to systematically investigate and validate patients with concurrent ALS and MS/MG/IP/DMPM according to ICD-codes at a national level and over a long period of time, resulting in the largest study to date on this topic. We further showed that patients with this confirmed overlap differed in terms of some clinical characteristics of ALS from patients diagnosed with only ALS. Patients diagnosed with concurrent ALS and MS/MG/IP/DMPM had an older age at first symptoms onset of ALS and a higher prevalence of bulbar onset. Bulbar onset of ALS is associated with a peculiar distribution of pathological findings in the extra motor cortical regions and the presence of neurofibrillary tangles and basophilic inclusions (Citation27). We also found differences between these two groups of patients in terms of care, such as less frequent use of Riluzole and non–invasive ventilation use among patients with concurrent diagnoses, which might be signs of a slower diseases progression or a less involvement of respiratory symptoms in ALS patients with concurrent neuroinflammatory disorders. The lower use of noninvasive ventilation might also be due to the higher prevalence of ALS patients with bulbar onset that are less likely to tolerate noninvasive ventilation than patients with limb onset (Citation28). Despite the reported differences might be partially due to a cohort effect, all patients diagnosed with concurrent ALS and MS/MG/IP/DMPM were diagnosed with ALS after 1997, when noninvasive ventilation treatment was widely practiced (Citation29), and Riluzole was approved in Sweden by the Medical Products Agency (Citation30).
The older age at symptoms onset may serve as an argument that if a link between neuroinflammation around the motor unit and ALS exists, there is a relatively long lag phase before onset of motor neuron degeneration. However, given the small numbers of patients with validated concurrence, the risk, if it exists, is modest. On the other hand, we also identified four and two patients diagnosed with MG and MS, respectively, after ALS. There is an important distinction between the two conditions, since symptoms of MG usually prompts investigations with a short diagnostic delay, while a proportion of MS patients are diagnosed with a long delay. We therefore speculate that in a small proportion of ALS patients, the neurodegenerative process triggers an autoimmune reaction against neuromuscular end plate proteins, as previously suggested (Citation31). In contrast, neuroradiological findings in the two patients diagnosed with MS after ALS indicated a preexisting, but un-diagnosed condition. Cellular and humoral autoimmune responses triggered by nerve injury are well recognized, but likely much more frequent with acute injuries compared to chronic neurodegenerative conditions (Citation32).
It is important to highlight that the shorter life expectancy of ALS patients greatly reduced the time at risk to develop MS/MG/IP/DMPM following ALS onset. This could partially explain the younger age at ALS symptoms onset and the longer survival among patients with MS diagnosed after ALS. Moreover, ALS symptoms might mask symptoms of other neuroinflammatory disorders leading to a potential underestimation of the concurrence between ALS and MS/MG/IP/DMPM.
Data validation
Our nationwide medical records review revealed that most of the patients with registered concurrence of ALS and MS/MG/IP/DMPM in the Swedish Patient Register had been misdiagnosed, calling for caution to use administrative healthcare databases in investigating concurrence between these diseases with similar symptomology. However, the high percentage of misdiagnosed ALS patients in this study is not representative of the accuracy of all ALS diagnoses in the Swedish Patient Register. Indeed, when we previously validated the register-based diagnosis for all ALS patients in Stockholm during 2013–2014 we found a positive predictive value of 91% (Citation33), which increased to 97% when including PLS (Citation26).
Strengths and limitations
The main strengths of our study are the nationwide design and the diagnostic accuracy decisions conducted by three experienced neurologists based on original medical records data. The fact that we were able to evaluate 92% of the medical records of all eligible patients identified during the study period provides evidence that our results are generalizable to the entire Swedish population.
Because genetic testing was not used widely during the entire study period in Sweden we lacked information about genetic characteristics of ALS. We were therefore unable to test whether patients with a validated concurrent ALS and MS/MG/IP/DMPM diagnosis had a particular genetic makeup compared to patients with only ALS. A meta-analysis of genome-wide association studies and polygenic analyses failed to provide evidence for an overlap in genetic susceptibility between MS and ALS (Citation34), but the study by Ismail et al, indeed found the C9ORF72 expansions in 80% of the seven patients diagnosed with ALS and MS.
We were also unable to compare whether functional impairment or staging of the disease differed between patients with a validated concurrent ALS and MS/MG/IP/DMPM and patients with only ALS, as during the study period information on ALS functional status and staging was not systematically recorded. As such information is available in the recently established Swedish Motor Neuron Disease Quality Registry (Citation26), there will be future possibilities to address this question in Sweden.
Moreover, we identified patients with a confirmed overlap from hospitals all over Sweden, but we compared them to patients with only ALS that were cared for in Stockholm. Hence, we cannot exclude the possibility that these two patient groups differed in terms of socioeconomic status and genetics.
Despite a nationwide effort with a study period of 23 years, due to the relative rarity of ALS and the even rarer concurrence of ALS with other neuroinflammatory disorders, we had only access to a limited number of patients with ALS a confirmed overlap.
Finally, we were unable to compare the number of observed patients with concurrent ALS and MS/MG/IP/DMPM with the number of expected ALS patients in the Swedish population diagnosed with MS/MG/IP/DMPM, or compare the clinical characteristics of patients with concurrent ALS and MS/MG/IP/DMPM to that of patients with MS/MG/IP/DMPM only.
Although we hypothesized a potential causal relationship between neuroinflammation around the motor unit and ALS, the fact that we found such a limited number of cases of overlap might also be due to chance alone. A concurrence of different diseases might also be attributable at least to some extent to surveillance bias, i.e. individuals with one medical condition might be more likely to receive a diagnosis of another medical condition due to their greater access to health care. Larger sample sizes, possibly through pooling together patients from different populations, might provide further insights and reveal if patients with concurrent ALS and MS/MG/IP/DMPM are different from patients with only ALS in terms of other clinical characteristics such as survival.
Conclusions
The concurrence of ALS and MS/MG/IP/DMPM is largely due to diagnostic uncertainty. In a small subgroup of ALS patients with defined clinical characteristics, motor neuron degeneration is triggered by a preceding neuroinflammation around the motor unit. However, we cannot exclude the possibility that the limited number of patients with confirmed biological overlap between ALS and MS/MG/IP/DMPM is due to chance alone. In contrast, a small number of patients presented almost simultaneously with ALS and MG, providing some support for previous data suggesting a link between degeneration of motor neurons and triggering of autoimmunity against end plate proteins.
Acknowledgements
We are grateful for the help received from thehospitals that provided us with the requested medical records and would also like to thank Amanda Regodón Wallin for her contribution to the data extraction of the general Swedish ALS patients used as comparison group in this study.
Disclosure statement
Dr. Fredrik Piehl has received research grants from Biogen, Genzyme, Merck KGaA, and Novartis, and fees for serving as Chair of DMC in clinical trials with Parexel, outside the submitted work. Dr. Elisa Longinetti, Dr. Olafur Sveinsson, Dr. Rayomand Press, Dr. Weimin Ye, Dr. Caroline Ingre, and Dr. Fang Fang report no disclosures.
Additional information
Funding
References
- Eisen A. Amyotrophic lateral sclerosis: a 40-year personal perspective. J Clin Neurosci. 2009;16:505–2.
- Ismail A, Cooper-Knock J, Highley JR, Milano A, Kirby J, Goodall E, et al. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J Neurol Neurosurg Psychiatry. 2013;84:79–87.
- Cui C, Longinetti E, Larsson H, Andersson J, Pawitan Y, Piehl F, et al. Associations between autoimmune diseases and amyotrophic lateral sclerosis: a register-based study. Amyotroph Lateral Scler Frontotemporal Degener 2020;22(3–4):211–9.
- Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology 2013;81:1222–5.
- Hemminki K, Li X, Sundquist J, Sundquist K. Familial risks for amyotrophic lateral sclerosis and autoimmune diseases. Neurogenetics 2009;10:111–6.
- Etemadifar M, Abtahi SH, Akbari M, Maghzi AH. Multiple sclerosis and amyotrophic lateral sclerosis: is there a link? Mult Scler. 2012;18:902–4.
- Tai H, Cui L, Guan Y, Liu M, Li X, Huang Y, et al. Amyotrophic lateral sclerosis and myasthenia gravis overlap syndrome: a review of two cases and the associated literature. Front Neurol. 2017;8:218.
- Gotaas HT, Skeie GO, Gilhus NE. Myasthenia gravis and amyotrophic lateral sclerosis: a pathogenic overlap. Neuromuscul Disord. 2016;26:337–41.
- Del Mar Amador M, Vandenberghe N, Berhoune N, Camdessanche JP, Gronier S, Delmont E, et al. Unusual association of amyotrophic lateral sclerosis and myasthenia gravis: a dysregulation of the adaptive immune system? Neuromuscul Disord. 2016;26:342–6.
- Restivo DA, Bianconi C, Ravenni R, De Grandis D. ALS and myasthenia: an unusual association in a patient treated with riluzole. Muscle Nerve. 2000;23:294–5.
- de Pasqua S, Cavallieri F, D’Angelo R, Salvi F, Fini N, D’Alessandro R, et al. Amyotrophic lateral sclerosis and myasthenia gravis: association or chance occurrence? Neurol Sci. 2017;38:441–4.
- Naik KR, Saroja AO, Mahajan M. Unusual occurrence of amyotrophic lateral sclerosis in myasthenia gravis. Amyotroph Lateral Scler. 2012;13:477–8.
- Yamashita S, Fujimoto A, Mori Y, Hirahara T, Mori A, Hirano T, et al. Coexistence of amyotrophic lateral sclerosis and myasthenia gravis. J Neuromuscul Dis. 2014;1:111–5.
- Ohnari K, Okada K, Higuchi O, Matsuo H, Adachi H. Late-onset myasthenia gravis accompanied by amyotrophic lateral sclerosis with antibodies against the acetylcholine receptor and low-density lipoprotein receptor-related protein 4. Intern Med. 2018;57:3021–4.
- Tseng CC, Chang SJ, Tsai WC, Ou TT, Wu CC, Sung WY, et al. Increased incidence of amyotrophic lateral sclerosis in polymyositis: a nationwide cohort study. Arthritis Care Res (Hoboken). 2017;69:1231–7.
- Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450.
- Ludvigsson JF, Otterblad-Olausson P, Pettersson BU, Ekbom A. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. Eur J Epidemiol. 2009;24:659–67.
- Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Finegan E, Chipika RH, Shing SLH, Hardiman O, Bede P. Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Frontotemporal Degener 2019;20(3–4):133–145.
- McNicholas N, Hutchinson M, McGuigan C, Chataway J. 2017 McDonald diagnostic criteria: a review of the evidence. Mult Scler Relat Disord. 2018;24:48–54.
- Huijbers MG, Niks EH, Klooster R, de Visser M, Kuks JB, Veldink JH, et al. Myasthenia gravis with muscle specific kinase antibodies mimicking amyotrophic lateral sclerosis. Neuromuscul Disord. 2016;26:350–3.
- Okuyama Y, Mizuno T, Inoue H, Kimoto K. Amyotrophic lateral sclerosis with anti-acetylcholine receptor antibody. Intern Med. 1997;36:312–5.
- Van den Bergh PY, Hadden RD, Bouche P, Cornblath DR, Hahn A, Illa I, et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. Eur J Neurol. 2010;17:356–63.
- European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–first revision. J Peripher Nerv Syst 2010;15:295–301.
- Asbury AK. Diagnostic considerations in Guillain-Barré syndrome. Ann Neurol. 1981;9:1–5.
- Longinetti ER, Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi LO, et al. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(7–8):528–37.
- Shellikeri S, Karthikeyan V, Martino R, Black SE, Zinman L, Keith J, et al. The neuropathological signature of bulbar-onset ALS: a systematic review. Neurosci Biobehav Rev. 2017;75:378–92.
- Gruis KL, Brown DL, Schoennemann A, Zebarah VA, Feldman EL. Predictors of noninvasive ventilation tolerance in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2005;32:808–11.
- Simonds AK. Home ventilation. Eur Respir J Suppl. 2003;47:38s–46s.
- Nygren I, Antonova K, Mattsson P, Askmark H. The ALS/MND prevalence in Sweden estimated by riluzole sales statistics. Acta Neurol Scand. 2005;111:180–4.
- Smeltzer JS. The effects of liability fears. N Engl J Med. 1989;320:1221.
- Javidi E, Magnus T. Autoimmunity after ischemic stroke and brain injury. Front Immunol 2019;10:686.
- Mariosa D, Hammar N, Malmstrom H, Ingre C, Jungner I, Ye W, et al. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: a more than 20-year follow-up of the Swedish AMORIS cohort. Ann Neurol. 2017;81:718–28.
- Goris A, van Setten J, Diekstra F, Ripke S, Patsopoulos NA, Sawcer SJ, et al. No evidence for shared genetic basis of common variants in multiple sclerosis and amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23:1916–22.