
1. Background information
It is just over 50 years since classical homocystinuria was first described independently in 1962 by Carson and Neill in Northern Ireland and Gerritsen and colleagues in Madison, Wisconsin [Citation1,Citation2]. In both cases, patients were studied because of mental retardation. Classical homocystinuria is caused by a deficiency of the enzyme cystathionine β-synthase deficiency (CBS) (EC 4.2.1.22) (see ). It is a rare inborn error of sulfur amino acid metabolism, characterized biochemically by elevation of plasma homocyst(e)ine and methionine and clinically by abnormalities variably affecting the eye (typically dislocated lenses), blood vessels (thromboembolism), central nervous system (intellectual deficits or psychiatric disorder), and the skeleton. Skeletal abnormalities include a Marfanoid habitus, genu valgus or varus, sternal deformity, and what has been described as osteoporosis. There is a wide range of severity. The pathophysiology appears mainly secondary to the elevated homocysteine levels. About half of all patients are biochemically responsive to the cofactor, given as oral pyridoxine (vitamin B6), with significant lowering of plasma homocysteine levels. These patients are more mildly affected, and sometimes asymptomatic throughout childhood and beyond. Pyridoxine-nonresponsive patients need other homocysteine-lowering treatment including low-methionine diet and betaine [Citation3]. Homocysteinemia and homocystinuria also occur in several inborn errors affecting homocysteine remethylation as well as in vitamin B12 and folic acid deficiencies and in renal insufficiency, but this commentary relates only to CBS deficiency.
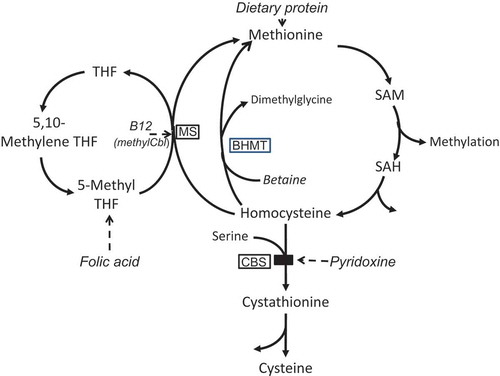
Figure 1. A simplified pathway of methionine metabolism and homocysteine remethylation.
Treatment compounds are shown in italics (but note that betaine is also naturally synthesised from choline). Enzymes are enclosed in boxes. BHMT – betaine-homocysteine methyltransferase; methylCbl – methylcobalamin; CBS – cystathionine beta-synthase; MS – methionine synthase; SAH – S-adenosylhomocysteine; SAM – S-adenosylmethionine; THF – tetrahydrofolate.
2. European guidelines for diagnosis and management
As for most other rare inborn errors, the pathophysiology is incompletely understood, diagnosis can be difficult, and evidence about exact treatment is modest. These issues have been taken up in the newly available ‘Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency’ [Citation4]. This guideline is one of four developed under the umbrella of eHOD – the acronym for the European Network and Registry for Homocystinurias and Methylation Defects [Citation5]. The stated aim of eHOD is to improve the health of subjects of all ages affected with these rare disorders. The content of this clear and extensive guideline seems to ensure achievement of this aim for CBS deficiency. It is well researched, well ordered, evidence based, and wide ranging. There are sections on clinical presentation and differential diagnosis, biochemical testing and problems relating to this, confirmatory testing by enzymology or mutation analysis, and genotype/phenotype correlations. There is a section on newborn screening – which is an important but currently imperfect route to diagnosis and particularly unsuitable for those patients who are pyridoxine responsive. All this necessary background leads up to the consideration of therapy, and the appropriate therapeutic targets for this disorder. Finally, there are sections on interventions for the various complications, and special management issues such as anesthesia, and fertility and pregnancy issues. In the guideline, each of the 41 statements, basically recommendations, is given with an introductory paragraph to encapsulate the whole, and a grading of the evidence supporting the recommendation.
2.1. General aims of treatment
The classic treatment of an inborn error due to an enzyme deficiency is to limit intake or formation of substrate (i.e. before the block), supply extra of the product (after the block), remove toxic metabolites if relevant, and use strategies to increase the relevant enzyme activity. In the case of CBS deficiency, homocysteine and methionine accumulate and plasma levels of cystathionine and products further down the metabolic pathway, mainly cysteine, can be deficient. These general ideas have been used in the early attempts to treat CBS deficiency. The main aim has been to keep the plasma homocysteine as close to the normal range as possible. The guideline notes that early studies in Ireland, Britain, and Australia showed that ‘good compliance with dietary treatment prevented ectopia lentis, osteoporosis, and thromboembolic events… and led to normal intelligence.’ They especially note that early diagnosis is crucial, and for those diagnosed early, for example, by newborn or family screening, prevention of all complications seems a realistic aim. Those diagnosed clinically will have presented with one of the complications; the aim then is to prevent further complications.
2.2. Lowering plasma total homocysteine: long-term data
The major aim of treatment is to lower the plasma total homocysteine (tHcy) level into the normal range. Evidence supporting this came early [Citation6], but the most compelling evidence is from small Irish cohort studies derived from largely newborn-screening-detected patients, who were pyridoxine-nonresponsive [Citation7]. Most of these patients were homozygous for the severe ‘Celtic’ mutation, p.Gly307Ser. Those compliant with treatment maintained plasma free homocysteine (fHcy) less than 11 µmol/L and remained complication free at 20–25 years of age. Unpublished information referred to in the guideline takes this timeline up to 43 years of age for compliant patients. This was not so for noncompliant or for late-treated patients. The guideline is clear about the measurement of plasma homocyst(e)ine levels and notes that the only available measure early on was of fHcy, which has major pre-analytical issues and is also insensitive at lower levels. From the late 1990s, most measures have been of tHcy (i.e. bound plus free homocysteine). Unfortunately these two do not have a stable relationship. As there are no long-term data relating outcome to tHcy levels, a plasma fHcy of <11 µmol/L is the level for which there is evidence of effectiveness of treatment. This is approximately equivalent to 120 μmol/L of tHcy (reference range c. 10–15 μmol/L). To develop robust treatment protocols, one must collect long-term data to be able to evaluate the effectiveness of any treatment, and this is one of the aims of e-HOD [Citation5].
2.3. Strategies for lowering plasma homocysteine
For pyridoxine-responsive patients, this is achieved using oral pyridoxine in pharmacological doses, plus folate, and sometimes vitamin B12. For those nonresponsive, a low-methionine diet including a methionine-free amino acid formula is used plus the addition of betaine, which lowers homocysteine via a B12/folate-independent remethylating pathway (). All of these strategies are individually well discussed in the guideline. In essence, the guideline suggests the following targets for plasma tHcy:
For pyridoxine-responsive patients <50 μmol/L. This is usually achievable, but if plasma levels stay above 50 μmol/L other medication can be considered, such as diet and betaine.
For pyridoxine nonresponsive patients <100 μmol/L (because of the evidence cited above, and the difficulty of achieving lower levels).
3. Expert opinion
For rare diseases, there is seldom any high-quality evidence about the effects of treatment and often only an imperfect notion of the pathophysiology. This is certainly true for CBS deficiency. In these cases, guidelines present a difficulty. They may represent simply the most usual approach to diagnosis and management and may perhaps be wrong, but become ‘set in stone’ before high-quality evidence to support them becomes available. The authors of these European guidelines have taken pains to avoid this as far as possible. They say in conclusion: ‘These guidelines are based on the best data currently available…’ and ‘should be used to identify areas in which data are lacking and to facilitate the design of international collaborative studies.’
The only therapeutic target identified is lowering plasma homocysteine levels. The evidence base supports this for CBS, and it is readily measureable, an important consideration. (By contrast, mild hyperhomocysteinemia is associated with neurodegenerative and cardiological diseases, but the mechanisms are different and there is no evidence from well-conducted trials that interventions to lower these homocysteine levels gives benefit, unlike the situation with CBS deficiency [Citation8]). In the future, other therapeutic targets may be found. Some treatment recommendations are without evidence but seem sensible, such as provision of cysteine supplementation. But this is complex because plasma cysteine is invariably lowered when circulating homocysteine is elevated, mainly due to competition for binding to plasma proteins. Tissue levels of cysteine have seldom been measured, and the effect of supplemental cysteine on tissue levels is unknown. In rats, dietary cysteine increases hepatic betaine-homocysteine methyltransferase, but decreases cystathionine synthase activity [Citation9]. Very little is said in the guideline about folic acid and B12 (except to recommend supplementation and monitoring, respectively), although these vitamins are vital for remethylation via the folate pathway. Low levels of either inhibit the remethylation pathway, and elevated homocysteine levels may increase utilization due to increased remethylation, thus making both more essential [Citation10]. It seems possible that regular B12 supplementation even when circulating levels fall within the normal range (not recommended in the guidelines) may enhance homocysteine lowering, but this has not been well investigated. And while the guidelines do recommend betaine as treatment, the authors appear surprisingly unenthusiastic. Certainly, it is correct to say that betaine alone is seldom sufficient treatment, but as an adjunct to a low-methionine diet in pyridoxine-nonresponsive patients it has been strikingly successful in allowing some relaxation of the diet. An extremely strict diet can prove difficult or impossible for many families. Treatment at once became easier when betaine use was first promulgated, both for the B6 nonresponsive and the moderately responsive patients [Citation11–Citation13]. A further area that needs more research is newborn screening. Currently, it is likely that good screening programs diagnose almost all of the pyridoxine-nonresponsive patients but few if any of the pyridoxine-responsive cohorts, who are thus at risk of missing out on the prevention of all the known complications in this complex and fascinating disorder [Citation14].
Despite these concerns, the new European guidelines for diagnosis and management of CBS are extremely useful and well researched, providing a sound basis for management. This initiative is greatly to be commended.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Carson NAJ, Neill DW. Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch Dis Childh. 1962;37:505–509.
- Gerritsen T, Vaughn JG, Waisman HA. The identification of homocystine in the urine. Biochem Biophys Res Com. 1962;9:493–496.
- Mudd S, Levy H, Kraus JP. Disorders of transsulphuration. Scriver CR, Beaudet A, Sly WS, et al., eds. The metabolic and molecular bases of inherited disease. 8th. New York (NY): McGraw-Hill; 2001; p. 2016–2040.
- Morris AAM, Kozich V, Santra S, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017. [Epub ahead of print].
- The European Network and Registry for Homocystinurias and Methylating defects. European Commission Health Programme. [Cited 2016 Oct 23]. Available from: https://www.e-hod.org/
- Mudd SH, Skovby F, Levy HL, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985;37(1):1–31.
- Yap S, Naughten E. Homocystinuria due to cystathionine beta-synthase deficiency: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis. 1998;21(7):738–747.
- Selhub J. Public health significance of elevated homocysteine. Food Nutr Bull. 2008;29(2 Suppl):SS116–SS125.
- Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost. 2000;26:219–225. Review.
- Blom HJ, Smulders Y. Overview of homocysteine and folate metabolism. J Inherit Metab Dis. 2011;34(1):75–81.
- Smolin LA, Benevenga NJ, Berlow S. Use of betaine for the treatment of homocystinuria. J Pediatr. 1981;99(3):467–472.
- Wilcken DE, Wilcken B, Dudman NP, et al. Homocystinuria – the effects of betaine in the treatment of patients not responsive to pyridoxine. 1983;309(8):448–453.
- Wilcken DE, Dudman NP, Tyrrell PA. Homocystinuria due to cystathionine beta-synthase deficiency – the effects of betaine treatment in pyridoxine-responsive patients. N Engl J Med. 1985;34(12):1115–1121.
- Huemer M, Kozich V, Rinaldo P, et al. Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. J Inherit Metab Dis. 2015;38(6):1007–1019.