
Abstract
Tight Junctions (TJ) create a paracellular barrier that is compromised when nonsteriodal anti-inflammatory drugs (NSAIDs) injure the gastric epithelium, leading to increased permeability. However, the mechanism of NSAID-induced gastric injury is unclear. Here, we examined the effect of indomethacin on barrier function and TJ in gastric MKN-28 cells. In concentration response studies, 500 µm indomethacin induced a significant decrease in transepithelial resistance (TER; 380 vs. 220 Ω·cm2 for control and indomethacin-treated cells respectively, p < 0.05), and increased dextran permeability by 0.2 vs 1.2 g/l (p < 0.05). These changes in barrier function were completely ameliorated by the p38 MAPK inhibitor (SB-203580) but not by JNK inhibitor (SP-600125) or MEK/ERK inhibitor (PD-98059). SiRNA knock down of p38 MAPK prevented the loss of barrier function caused by indomethacin in MKN-28 cells. Western analyses of TJ proteins revealed that expression of occludin was reduced by indomethacin, whereas there was no change in other TJ proteins. The loss of occludin expression induced by indomethacin was prevented by inhibition of p38 MAPK but not JNK or ERK and also by siRNA of p38 MAPK. Immunofluorescence revealed disruption of occludin localization at the site of the tight junction in indomethacin-treated cells, and this was attenuated by inhibition of p38 MAPK. NSAID injury to murine gastric mucosa on Ussing chambers revealed that indomethacin caused a significant drop in TER and increased paracellular permeability. Pretreatment with the p38 MAPK inhibitor significantly attenuated the disruption of barrier function, but JNK and MEK/ERK inhibition had no effect. Western blot analysis on gastric mucosa reveled loss of TJ protein occludin by indomethacin, which was prevented by inhibition of p38 MAPK. This data suggests that indomethacin compromises the gastric epithelial barrier via p38 MAPK inducing occludin alterations in the TJs.
KEYWORDS:
Introduction
Use of nonsteroidal anti-inflammatory Drugs (NSAIDs) is known to cause gastrointestinal injury.Citation1,3,10 In particular, the possibility of increased risk of gastrointestinal injury with high concentrations and chronic administration of NSAIDs for treatment of pain is a major concern. Various factors, including reduction in prostaglandins, increases in acid secretion, reduction in mucus secretion, disturbances in mucosal pH, increased production of oxygen free radicals, and compromises in mucosal barrier function have been found to play a role in the development of NSAID-induced gastric injury.Citation32,34,35 However, the effects of indomethacin on the gastric epithelial tight junction (TJ) barrier are not well studied. TJs are the most apical component of intercellular junctional complexes and are composed of occludin, members of the claudin family of proteins, and junctional adhesion molecule.Citation30 In gastric barrier function, besides TJ, mucus and endogenous prostaglandins also play a role in preventing mucosal injury and back diffusion of acid into the tissues. In the gastric mucosa, NSAIDs are known to cause mucosal injury in a cyclooxygenase (COX)-dependent and independent manner. The NSAID indomethacin is also known to induce apoptosis in the gastric mucosa.Citation13,31 Experimentally, H.pylori, an organism involved in gastric ulceration has been shown to damage the gastric barrier by disrupting tight junctions.Citation36 However, the relationship between gastric ulcer pathogenesis, apoptosis, and TJ barrier disruption is unclear.
Mitogen-activated protein kinases (MAPK) respond to extracellular stimuli and play a critical role in regulating cellular processes including proliferation, differentiation, apoptosis, and gene expression. The role of MAPKs in the regulation of the TJ barrier has been studied in various cell lines and tissues.Citation5,14,16,26 Recently, it has been shown that the NSAID aspirin injures the gastric barrier by activating p38 MAPK, with resultant loss of claudin-7.Citation20 Other than this report, there is a paucity of information on the role of MAPKs in the gastric epithelial TJ barrier.
The goal of the present study was to determine if MAPKs play a role in indomethacin-induced changes in the gastric barrier. The results of our study show that indomethacin induces gastric barrier dysfunction in MKN-28 human gastric epithelial cells. Indomethacin induces loss of the TJ protein occludin. Inhibition of p38 MAPK prevented indomethacin-induced loss of barrier function and disruption of occludin. JNK and ERK inhibition had no effect on TJ barrier function. In ex vivo studies, indomethacin-induced barrier dysfunction via the tight junction protein occludin in murine gastric mucosa, which was prevented by inhibition of p38 MAPK.
Results
Indomethacin reduces epithelial barrier function of MKN-28 cells
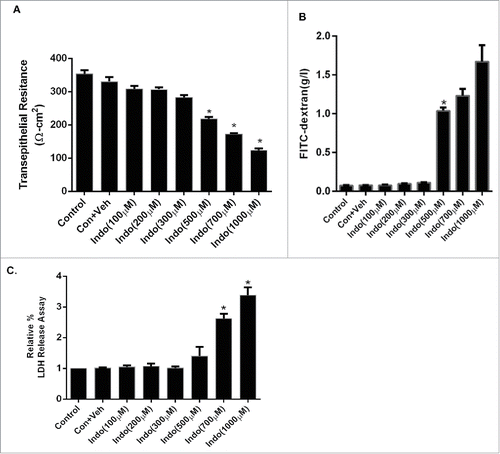
Indomethacin at 100 µM-300 µM did not reveal any significant drop in the TER or an increase in epithelial permeability, whereas indomethacin at concentrations of 500 µM-1000 µM caused a concentration-dependent and significant drop at 24 hours after exposure. Consistent with the TER data, indomethacin at concentrations of 100 µM-300 µM had no effect on FITC dextran flux, whereas concentrations of 500 μM-1000 μM caused significant increases in the FITC dextran flux at 24 hours post exposure (). Similarly, There was no evidence of any cytotoxic effect of indomethacin at the concentration of 100-500 µM, with up to 24 hours exposure based on LDH release assays (), whereas concentrations of 700 µM-1000 µM caused significant toxicity. Collectively, based on these findings, the 500 µM concentration of indomethacin was used in further studies.
Figure 1. Concentration response curve for indomethacin-induced loss of barrier function in the MKN-28 cell line. (A) Indomethacin (100 µM-1 mM) decreased TER in a concentration-dependent manner. Indomethacin concentrations of 500 µM, 700 µM, and 1 mM reduced the TER significantly at 24-hours post-treatment. *p < 0.05. (B) Indomethacin (100 µM-1 mM) increased the FITC dextran flux in a concentration dependent manner. Indomethacin concentrations of 500 µM, 700 µM, and 1 mM increased FITC dextran flux significantly at 24-hours post-treatment. *p < 0.05. (C) Examination of indomethacin-induced cell toxicity by LDH release assay. Indomethacin concentrations of 700 µM, and 1 mM caused significant increase in LDH release 24-hours post-treatment, whreas there was no increase in LDH in cells exposed to 500 µM over the same time period. *p < 0.05. These data are represented as the average of more than 3 identically treated monolayers, from 3 independent experiments.
Effects of MAPK inhibitors on indomethacin-induced decreases in barrier function
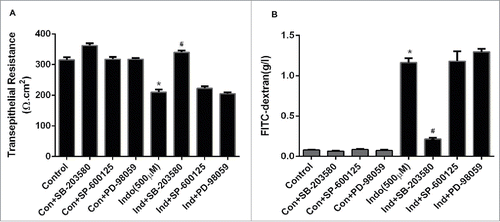
In the next set of experiments, we studied the effects of MAPK inhibitors on indomethacin-induced decreases in TER. Indomethacin (500 µM) reduced TER significantly at 24 hours post-treatment, while pre-treatment of cell monolayers with the p38 MAPK inhibitor SB-203580 (10 µM) significantly reduced the drop in TER caused by indomethacin with up to 24 hours of exposure (p < 0.05; ). However, the JNK inhibitor SP-600125 (10 µM) and ERK inhibitor PD-98059 (10 µM) did not have any effect on indomethacin-induced decreases in TER (). Treatment of MKN-28 cell monolayers with PD-98059 (10 µM), SB-203580 (10 µM), or SP-600125 (10 µM) alone (for 24 hours) did not influence the baseline TER (). Pretreatment of cell monolayers with the p38 MAPK inhibitor SB-203580 (10 µM) significantly prevented the increase in FITC-dextran flux caused by indomethacin (). Pretreatment with the JNK inhibitor SP-600125 (10 µM) and the MEK inhibitor PD-98059 (10 µM) had no effect on indomethacin-induced increases in epithelial permeability. Furthermore, the MAPK inhibitors PD-98059 (1-10 µM), SB-203508 (1-10 µM), or SP-600125 (1-10 µM) alone (for 24 hours) did not have any significant effects on gastric epithelial permeability. Thus the TER and FITC dextran permeability data clearly indicated that p38 MAPK, but not JNK and ERK, are involved in the indomethacin-induced gastric cell barrier dysfunction.
Figure 2. Effect of MAPK inhibition on indomethacin-induced changes in the epithelial barrier in MKN-28 cells. (A) Indomethacin (500 μM) reduced the TER significantly at 24-hours post treatment, while inhibition of p38 MAPK and JNK significantly attenuated the drop in TER caused by indomethacin (*p < 0.005). (B) Indomethacin (500 µM) caused a significant increase in the paracellular permeability of dextran (4KD) in MKN-28 cells (24-hours post exposure), while consistent with effect on TER, p38 MAPK and JNK inhibition attenuated the indomethacin-induced increase in paracellular permeability (#p < 0.05 vs Indo alone). The data are represented as the average of more than 3 identically treated monolayers, from 3 independent experiments.
Indomethacin reduces the expression of the tight junction protein occludin
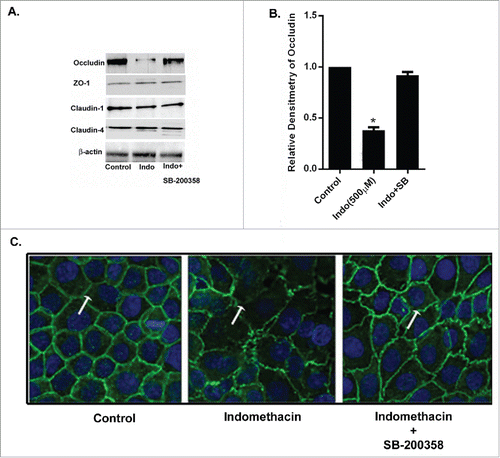
To further evaluate the effects of indomethacin on gastric cell barrier function, we examined the expression of select TJ proteins relevant to gastric epithelial barrier. There were no changes in the expression of ZO-1, claudin-1, claudin-4, or claudin-7 in total cell extracts after exposure to indomethacin (500 µM for 24 hours; ). Claudin-3 was not detected in MKN-28 cell lysates. The expression of occludin protein was significantly reduced after exposure to indomethacin (). Furthermore, the indomethacin-induced decrease in the expression of occludin was attenuated when cell monolayers were pretreated with the p38 MAPK inhibitor, SB-203580. Alternatively, pretreatment with SB-203580 (10 µM) alone did not change the expression of occludin. Indomethacin-induced decreases in the expression of occludin were not changed by the pretreatment with PD-98059 and SP-600125 alone (data not shown).
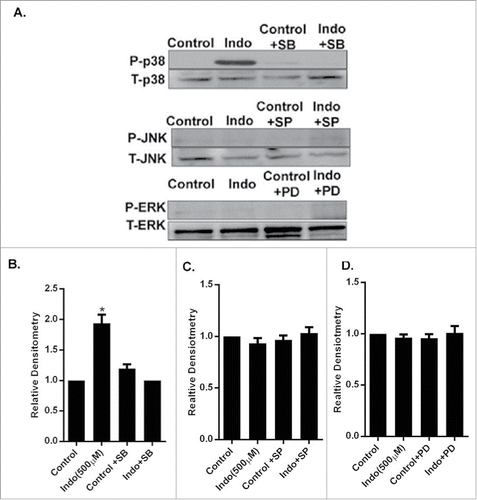
Figure 3. Evaluation of MAPK phosphorylation in MKN-28 cells in the presence of indomethacin. (A) Indomethacin-induced phosphorylation of p38 MAPK was blocked by pretreatment with the p38 MAPK inhibitor SB-203580. Similarly the indomethacin induced phosphorylation of JNK and ERK was also blocked by SP-600125 and PD-98059 respectively. Indomethacin-treated cells, though, showed phosphorylation of ERK, it was not significantly different from the control cells. (*p < 0.05 vs. control). (B) Relative densitometry of phosphorylated p38 MAPK as compared to total p38 MAPK. The data are represented as the average of more than 3 identically treated monolayers from 3 independent experiments. The data are presented the same way for the other MAPK analyses in parts C and D. (C) Relative densitometry of phosphorylated JNK as compared to total JNK. (D) Relative densitometry of phosphorylated ERK as compared to total ERK.
p38 MAPK but not JNK or ERK mediates indomethacin-induced disruption of occludin
We found that exposure of MKN-28 cell monolayers to indomethacin induced phosphorylation of p38 MAPK and not JNK and ERK. The indomethacin-induced phosphorylation of p38 MAPK was blocked by pretreatment with the p38 MAPK inhibitor, SB-203580 (). Similarly, indomethacin treatment did not induce any phosphorylation of JNK. The JNK inhibitor blocked JNK phosphorylation (). ERK MAPK was found to be phosphorylated in untreated MKN-28 cells, possibly because of the presence of serum in the culture media. Indomethacin-treated cells also showed phosphorylation of ERK, which was not significantly different from the control cells.
Figure 4. Indomethacin causes a decrease in occludin via activation of p38 MAPK. (A) Indomethacin exposure for 24 hours markedly reduced expression of occludin but not claudin-2, claudin-4 or ZO-1. Inhibition of p38 MAPK with SB-200358 attenuated the loss of expression of occludin caused by indomethacin. (B) Relative densitometry of occludin to control levels, confirming the significant decrease in occludin expression in the presence of indomethacin alone. Inhibition of p38 MAPK with SB-203580 significantly attenuated the reduced expression of occludin. (*p < 0.05 vs. control and Indo + SB). The data are represented as the average of more than 3 identically treated monolayers from 3 independent experiments. (C) MKN-28 cells after pretreatment with MAPK inhibitors for 1 hour were exposed to indomethacin (500 μM) for 24 hours. Cells were fixed and stained for occludin (green fluorescence) and examined by confocal microscopy. Immunolocalization of occludin at the tight junctions (arrow in control) was found to be disrupted in indomethacin exposed cells (500 µM) (arrow) while inhibition of p38 MAPK with SB-203580 prevented the loss of occludin at the TJs (arrow) 200X. The images are representative of more than 3 identically treated monolayers from 3 independent experiments.
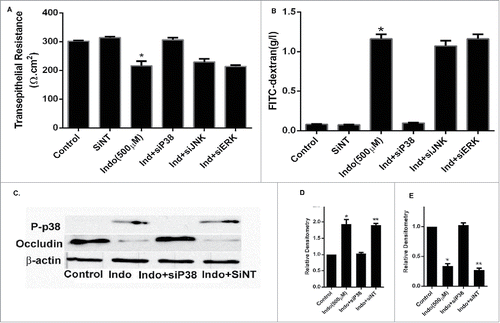
In further experiments, we studied morphological evidence for the role of MAPK in indomethacin-induced barrier dysfunction by evaluating immunolocalization of the TJ protein occludin. Confocal immunofluorescence revealed that indomethacin treatment disrupted localization of occludin at the tight junctions (, arrowheads). Furthermore, the indomethacin-induced disruption of occludin was attenuated by inhibition of p38 MAPK but not JNK (, asterisks show occludin in the cytoplasm) or ERK (). This data indicated that the epithelial barrier dysfunction caused by indomethacin was mediated by activation of p38 MAPK, which in turn compromised localization of the tight junction protein occludin. To further assess the involvement of p38 MAPK in the indomethacin effect on gastric barrier function via disruption of occludin, p38 MAPK expression was silenced in MKN- 28 monolayers by p38 siRNA transfection (). The siRNA induced knock-down of p38 MAPK also inhibited the indomethacin (500 µM) induced drop in MKN-28 cell TER () and increase in FITC- dextran flux across the MKN-28 Monolayers (). The transfection with scrambled siRNA did not affect the indomethacin-induced drop in MKN-28 TER or the increase in inulin flux. Furthermore, the p38 MAPK siRNA transfection inhibited the indomethacin (500 µM) induced decrease in occludin expression (). Together, these data suggested that indomethacin (500 µM) induced a reduction in tight junction protein occludin expression and increased MKN-28 TJ permeability via the p38 MAPK signaling pathway.
Figure 5. Effect of p38 siRNA on occludin in indomethacin-treated MKN-28 cells. (A) SiRNA of p38 MAPK prevented the decrease in transepithelial resistance caused by indomethacin (500 µM) when compared to control. Non-specific SiRNA did not have an appreciable effect on control cells The data are represented as the average of more than 3 identically treated monolayers from 3 independent experiments. (*p < 0.001 vs. control and Ind+siP38). (B) SiRNA of p38 MAPK attenuated the increase in inulin flux caused by indomethacin (500 µM) across the MKN-28 cells when compared to control. The data were generated in the same way as panel A. (*p < 0.001 vs. control). (C) Indomethacin caused a decrease in tight junction occludin protein expression. Knocking-down p38 MAPK by siRNA transfection prevented the indomethacin-induced decrease in occludin expression as assessed by western analyses. This blot is representative of the data shown in panels D and E. (D) Relative densitometry for phosphorylated p38 MAPK in the presence of indomethacin, and in cells transfected with siRNA for p38 MAPK. The data is represented as the average of more than 3 identically treated monolayers, from 3 independent experiments. (E) Relative densitometry of occludin in cells treated with indomethacin or indomethacin in cells transfected with siRNA for p38MAPK. The data are shown in the same way as panel D.
Ex-vivo Effect of MAPK inhibition on indomethacin-induced mucosal permeability in the gastric mucosa
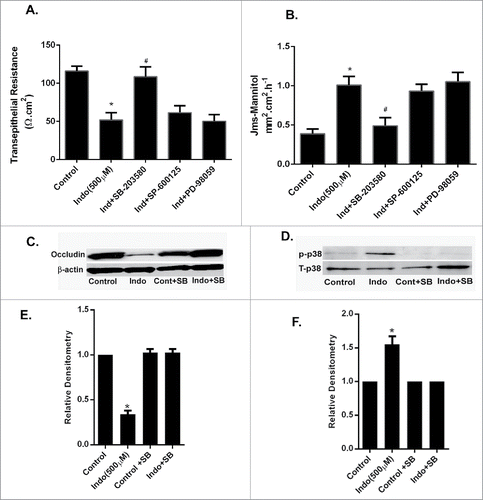
To determine if MAPK signaling is involved in indomethacin-induced gastric mucosal injury, we conducted ex vivo studies using murine gastric mucosa. The gastric mucosa was harvested from C57BL/6 wild type mice and was immediately mounted in Ussing chambers and exposed to indomethacin with or without pretreatment of MAPK inhibitors. Indomethacin (500 µM) caused a significant drop in the TER in murine gastric mucosa. Pretreatment with the P38 MAPK inhibitor and the JNK inhibitor significantly attenuated the indomethacin-induced drop in TER in the mucosa, but ERK inhibition had no effect (). Furthermore indomethacin also induced increases in the paracellular flux of mannitol in the gastric mucosa. Pretreatment with the p38 MAPK inhibitor SB-203580 or the JNK inhibitor SP-600125 significantly attenuated indomethacin-induced increases in mannitol flux, whereas ERK inhibition did not change the increased permeability caused by indomethacin (). Consistent with our in- vitro experiments, western blot studies of murine gastric tissues collected from the Ussing chambers revealed that the expression of occludin protein was significantly reduced after exposure to indomethacin for 90 minutes (). Similarly, indomethacin also induced phosphorylation of p38 MAPK (). Furthermore, the indomethacin-induced decrease in the expression of occludin was attenuated when the gastric tissues were pretreated with SB-203580. Treatment with SB-203580 (10 µM) alone did not change the expression of occludin. Indomethacin-induced decreases in the expression of occludin were not changed by pretreatment with PD-98059 or SP-600125 alone (data not shown).
Figure 6. Effect of MAPK inhibition on indomethacin-induced alterations in gastric barrier function. (A) The gastric mucosa from wild type C57/BL-6 mice (n = 6) was mounted on Ussing chambers. Indomethacin (500 µM) reduced the TER significantly (*p < 0.05, compared to control cells, #p < 0.05 compared to Indo+SB) while pretreatment with p38 MAPK and JNK inhibitors prevented the drop in TER caused by indomethacin. (B). In the Ussing chamber experiments, as in A, indomethacin caused a significant increase in paracellular permeability of mannitol in the gastric mucosa compared to untreated tissues (*p < 0.05, compared to control cells, #p < 0.05 compared to Indo+SB). In the gastric mucosa pretreated with p38 MAPK and JNK inhibitors, the mannitol permeability of indomethacin-treated tissues was comparable to that of control gastric mucosa. (C) Gastric tissues from Ussing chamber experiments were collected and subjected to western analyses. Indomethacin exposure for 90-minutes caused reductions in expression of occludin. (D) Western analysis of gastric tissues collected as in C showed an increase in the expression of phosphorylated p39 MAPK in the presence of indomethacin alone, which was markedly reduced by additional treatment with the p38 MAPK inhibitor SB-203580. (E) Relative densitometry of the n = 6 tissues western blotted for occludin expression (representative blot shown in C). Note the significant reduction in occludin expression caused by treatment with indomethacin (500 μM), which was restored by pre-treatment with the p38 MAPK inhibitor SB-203580 (*p < 0.05 vs. all other treatment groups). (F) Relative densitometry of the n = 6 tissues protein gel blotted for p38MAPK expression (representative blot shown in D). Note the significant increase in phosphorylated p38 MAPK in the presence of indomethacin (500 μM), which was reduced by pre-treatment with the p38 MAPK inhibitor (*p < 0.05 vs. all other treatment groups).
Discussion
Nonsteriodal anti-inflammatory drugs (NSAIDs) are the most widely used therapeutic agents for the treatment of pain, inflammation, and fever.Citation29 The anti-inflammatory action of NSAIDs is mediated through the inhibition of cyclooxygenase (COX). COX is an enzyme complex that initiates prostaglandin synthesis, and prostaglandins have a strong propensity for inducing inflammation. Prostaglandins, such as prostaglandin E2 (PGE2), inhibit apoptosis and stimulate cell growth and angiogenesis.Citation11 However, several lines of evidence suggest that gastropathy by NSAIDs also involves COX-independent mechanisms.Citation23,24 For instance, NSAIDs caused apoptosis and the inhibition of cell growth in COX null fibroblasts and tumor cells in which COX expression was absent.Citation9,38 Indomethacin, as well as other NSAIDs, induces gastric epithelial damage.Citation13,31 Understanding the mechanisms involved in indomethacin-induced gastric damage may provide new strategies for the prevention of NSAID-associated gastropathy. In the present study, we examined how paracellular junctional permeability in gastric epithelial cells and gastric mucosa responded to exposure to indomethacin in vitro and ex vivo. We found that indomethacin at a concentration of 500 µm reduced the TER in MKN-28 gastric epithelial cells and increased permeability to the paracellular probe FITC-Dextran and 3H-mannitol, respectively. Higher concentrations of indomethacin have been reported to be required to induce gastropathy in prior studies.Citation33 Other studies using oral administration of aspirin in rats have shown that the concentration in the stomach were 10–100 µM.Citation7 In our studies with indomethacin, we found that a 500 µM concentration of indomethacin induced significant decreases in TER and increase in permeability at 24 hours post treatment without any cytotoxic effects. To extrapolate to clinical medicine, using the upper limit of the oral dose (50 mg), this would equate to 131 µmol of indomethacin, but would potentially be less diluted once in the stomach than the experimental dose of indomethacin used in the present study (500 µmol in 1 L of diluent).Citation19
In further in vitro experiments we demonstrated that p38 MAPK and JNK are involved in indomethacin-induced loss of barrier function in the gastric cell line, and this loss of barrier occurs through loss of the TJ protein occludin. The protective effect of inhibition of p38 MAPK on tight junction protein occludin and thus on the mucosal barrier was further demonstrated in ex vivo studies using murine gastric mucosa. The role of intracellular signaling pathways in the regulation of TJ permeability has only recently begun to be elucidated. Studies published to date have focused largely on the MEK-ERK pathway. Specifically, it has been reported that growth factor-dependent activation of ERK1/2 causes an increase in TER associated with modulation of claudin expression in MDCK II cellsCitation6,15,28 as well as in T84 intestinal epithelial cells.Citation12 Although the involvement of p38 MAPK in the regulation of epithelial barrier function has been less thoroughly investigated, circumstantial evidence indicates that activation of this MAPK may increase TJ permeability. Thus, inhibition of p38 MAPK was reported to prevent the disruption of the TJ barrier induced by various stimuli in different epithelial cell types.Citation20,22 Similarly, inhibition of JNK signaling has been shown to reduce claudin-2 expression in MDCK II cells.Citation4,8 The role of MAPK in epithelial permeability has been emphasized in several studies. Activation of ERK1/2 induced H2O2-mediated permeability with rearrangement of the TJ protein occludin in endothelial HUVEC cells.Citation14 Similarly, the role of JNK in osmotic stress-induced barrier dysfunction in the intestinal epitheliumCitation25 and the role of ERK in the H2O2-mediated intestinal epithelial cell barrier dysfunction have been demonstrated.Citation27 In the gastric cell line MKN-28, Oshima et al., Citation20 has specifically shown that aspirin-induced epithelial barrier permeability occurred via activation of MAPK especially p38 MAPK changes in the tight junction protein claudin-7. Aspirin triggered phosphorylation of MAPKs, including p42/44 MAPK, p38 MAPK, and JNK. However, only the p38 MAPK inhibitor SB-203580 attenuated the aspirin-induced decrease in TER and aspirin-induced increase in permeability. Although the role of MAPKs in epithelial barrier function and TJ function appears to be clear, the underlying mechanism and involvement of specific MAPK components varies, possibly due to the cell type or the model used in experimental studies.
Our current study provided evidence that indomethacin induced disruption of tight junction protein occludin in MKN-28 cell line and this was prevented by pretreatment with p38 MAPK inhibitor SB-203580. Among several mechanisms by which indomethacin induce gastric mucosal damage,Citation31 activation of p38 MAPK is one of the most important components in the activation of endoplasmic reticulum stress and further damage.Citation21 In our studies, along with prevention of occludin disruption, inhibition of p38 MAPK prevented indomethacin-induced gastric damage shown by Transepithelial resistance. Recently it has been shown that apoptosis or caspase-3 activation required occludin in experimentally TNF-α-induced apoptosis.Citation37 On the other hand, occludin has been shown to be essential for transducing apoptotic signalsCitation37 and delocalized extrajunctional occludin forms a complex with the apoptotic machinery including death-inducing signaling complex (DISC), caspases-8 and -3, the death receptor FAS and the adaptor molecule FADD.Citation2 Thus it is possible that p38 MAPK inhibition prevents the downstream signaling for apoptosis where occludin may act as one of the signaling molecules inducing apoptosis, which requires further studies.
The present study shows that although indomethacin induces phosphorylation of p38 MAPK and indomethacin-induced reductions in TER and increased permeability was only attenuated by inhibition of p38 MAPK and not JNK or ERK. We also found that occludin expression and localization was preserved by inhibition of p38 MAPK but not JNK or ERK. Our current data support the idea that disruption of barrier function involves p38MAPK activation and a specific tight junction protein occludin is targeted by particular signaling components of the p38 MAPK pathways. Thus p38 MAPK could be a potential therapeutic target in preventing indomethacin induced mucosal damage to the gastric mucosa.
Materials and Methods
Cell line
MKN-28, a human gastric adenocarcinoma cell line, was a kind gift from Dr. Richard Peek, Vanderbilt University. The cell line was cultured using RPMI-1640 media (Sigma, St. Louis, MO), supplemented with 10% FBS, 2.5 mg/ml amphotericin B, and 20 μg/ml gentamicin. The culture medium was changed every 48-72 hours. MKN-28 cells were subcultured after partial digestion with 0.25% trypsin and EDTA in Ca2+ and Mg2+ - free PBS.
Reagents
The primary antibodies used were mouse anti-occludin, mouse anti- ZO-1, rabbit anti-claudin-2, rabbit anti-claudin-3, rabbit anti-claudin-4 (Invitrogen, Carlsbad, CA). Alexa 488-conjugated anti-mouse and Cy3-conjugated anti-rabbit IgG secondary antibodies were purchased from Invitrogen. Anti phospho-p38 MAPK, p42/44 MAPK, and JNK antibodies were purchased from Invitrogen. ERK inhibitor PD-98059, p38 MAPK inhibitor SB-203580 and JNK inhibitor (SP-600125) were obtained from Promega (Madison, WI). Indomethacin (Cat.No.I-7378) and DMSO were purchased from Sigma-Aldrich, St. Louis, Missouri, USA.
Determination of epithelial monolayer resistance and epithelial permeability
The epithelial voltohmeter (World Precision Instruments, Sarasota, FL) was used for measurments of the transepithelial electrical resistance (TER) of the filter grown MKN-28 gastric monolayers as reported earlier.Citation20 The epithelial paracellular permeability in response to indomethacin was determined by FITC-labeled dextran (FD-4, molecular mass: 4,000; Sigma) as a permeable probe.Citation17 Briefly, confluent MKN-28 cell monolayers on tissue culture inserts were washed twice with HBSS and a 500 µl aliquot of HBSS containing 5 mg/ml FITC dextran was added to the apical chamber. After incubation of cell monolayers at 37°C for 30 min, a 100 µl sample was taken from the lower chamber, and the absorbance of FITC-dextran was determined at 488 nm using a spectrophotometer. The permeability was determined either under control conditions, or after an exposure to indomethacin (100 µM-1 mM), and after pretreatment with PD (10 µM for 60 min), SB-203580 (10 µM for 60 min), or SP-600125 (10 µM for 60 min).
For all TER measurements, the inserts were plated at an equal density; the readings were taken in triplicate per monolayer and averaged. Upon reaching a reasonably stable resistance (> 300 Ω.cm2), the culture medium from the upper (apical) compartment of the monolayer was removed and replaced with medium containing different concentration of indomethacin (100 µM-1 mM) or control medium. After the concentration response study, a 500 µM concentration of indomethacin was used for all further experiments. In respective experiments, monolayers were pretreated with PD-98059 (10 µM for 60-min), SB-203580 (10 µM for 60-min), or SP-600125 (10 µM for 60-min) before the addition of indomethacin (500 µM). The data is represented as the average of more than 3 identically treated monolayers, from 3 independent experiments.
siRNA of p38 kinase
Targeted siRNAs were obtained from Dharmacon (Chicago, IL). MKN-28 monolayers were transiently transfected using DharmaFECT transfection reagent (Thermo Scientific, Lafayette, CO). Briefly, 5 × 105 cells per filter were seeded into a 12-well transwell plate and grown to confluency. MKN-28 cells monolayers were then washed twice with PBS and 0.5 ml Accell medium was added to the apical compartment of each filter and 1.5 ml was added to the basolateral compartment of each filter. Five nanograms of the siRNA of interest and 2 μl DharmaFECT reagents were added in Accell medium to the apical compartment of each filter. The Indomethacin (500 µM) experiments were carried out 96 h after transfection. The efficiency of silencing was confirmed by protein gel blot analysis.
Determination of lactate dehydrogenase (LDH) release for measuring cell toxicity
LDH activity in the medium, an index of cellular toxicity and death was measured using a spectrophotometric assay with pyruvate and NADH as the substrates (Pierce TM, LDH cytotoxicity assay kit). LDH release into the incubation medium was expressed as relative percentage of total LDH activity present at the beginning of incubation.
Immunofluorescence
For immunofluorescence of TJ proteins, confluent monolayers of MKN-28 cells on 1.2 cm diameter glass coverslips were exposed to different experimental treatments. Followed by wash in cold PBS, the coverslips were fixed with methanol (30-min) and stored at −20°C until further staining. Cover slips were stained for occludin, claudins, ZO-1, and activated Caspases-3 using appropriate dilutions of the primary and secondary antibodies and the nuclear stain. The coverslips were mounted in fluorescent mounting medium and examined with a Nikon Eclipse 2000E inverted microscope equipped with the Nikon C1 confocal laser scanning system. Examination of tissues was performed in a blinded fashion to ensure lack of bias.
Gel electrophoresis and western blot analysis
Gel electrophoresis and western blot analysis was performed as previously described.Citation17 Cell lysates and gastric tissue lysates were prepared by adding cell lysate buffer (50 mM Tris, 5 mM MgCl2·H20, 25 mM KCl, 2 mM EDTA, 40 mM sodium fluoride, 4 mM sodium orthovandate, 1% Triton X-100, and protease inhibitor cocktail (Roche)) directly to culture dish. The cells were scrapped, sonicated and centrifuged (10,000 RPM for 10 min) and the supernatant were stored at −80°C. All the samples from cell lysates and tissue extracts were subjected to protein analysis using BCA Protein Assay Kit (Pierce, Rockford, IL). Tissue or cell extracts (amounts equalized by protein concentration) were mixed with appropriate volumes of 2 × SDS-PAGE sample buffer and boiled for 4 min. Lysates were loaded on a 4–12% SDS polyacrylamide gradient gel, and electrophoresis was carried out according to standard protocols. Proteins were transferred to a PVDF membrane (Immobilon, Millipore, Billerica, MA) by using an electroblotting minitransfer apparatus (Biorad). Membranes were blocked at room temperature for 2-hours in 5% dry powdered milk in Tris-buffered saline plus 0.05% Tween 20 (TBST), and then incubated overnight in a primary antibody solution at 4°C. After washings in TBST, membranes were incubated with horseradish peroxidase conjugated secondary antibody, and the blots were developed for visualization of protein bands with luminol enhancer solution (Pierce, Rockford, IL). Densitometric analyses were performed using Image J software (NIH, https://imagej.nih.gov/ij/)
Ussing chamber studies
C57BL/6 wild type mice were obtained from a mouse colony maintained at NC State University. The mice were euthanized by an Institutional Animal Care and Use approved protocol, the entire stomach was clamped proximally and distally with Doyen intestinal forceps and placed in 10 µM indomethacin in oxygenated (95%O2/5% CO2) Ringer solution mM: 154 Na+, 6.3 K+, 137 Cl−, 0.3 H2PO4, 1.2 Ca2+, 0.7 Mg2+, 24 HCO3−, pH 7.4) to block prostaglandin production. The stomach was sharply incised at the lesser curvature, washed in Ringer solution, and placed in 0.14-cm2- aperture Ussing chambers.Citation18 The tissues were bathed on the serosal and mucosal sides with Ringer solution. The serosal bathing solution contained indomethacin (5 μM) and 10 mM glucose, which was osmotically balanced on the mucosal side with 10 mM mannitol. Bathing solutions were oxygenated (95% O2/5% CO2) and circulated in water-jacketed reservoirs maintained at 37°C. The spontaneous potential difference (PD) was measured with Ringer-agar bridges connected to calomel electrodes, and the PD was short circuited through AgCl electrodes with a voltage clamp that corrected for fluid resistance. Transepithelial electrical resistance (TER, Ω·cm2) was calculated from the spontaneous PD and short-circuit current. After the equilibration period of 15-min, the experiments were run for up to 120-min. Indomethacin (500 μM) was added on the mucosal side with or without pretreatment with MAPK inhibitors. In each Ussing chamber experiment, duplicate tissues were studied from each animal and more than 3 animals were studied in each experimental group. After the experiment gastric tissues were collected from the chambers and subjected for protein gel blot analysis.
Statistical analysis
Statistical significance of differences between mean values was assessed with Student's t-tests for unpaired data and ANOVA analysis whenever required. All reported significance levels represent 2-tailed P values. A P value of < 0.05 was used to indicate statistical significance. All experiments were repeated at least 3 times to ensure reproducibility.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are thankful to Dr. Richard Peek, Vanderbilt University for providing MKN-28 cell line. The authors are also thankful to Karen Young and Prashant Nighot, Department of Clinical Sciences, NCSU for technical assistance. The authors also thank Dr. Troy Ghashghaei, NCSU for assistance with confocal immunofluorescence microscopy.
Funding
This article was funded in part by the NC State College of Veterinary Medicine.
References
- Barrier CH, Hirschowitz BI. Controversies in the detection and management of nonsteroidal antiinflammatory drug-induced side effects of the upper gastrointestinal tract. Arthritis Rheum 1989; 32(7):926-32; PMID:2787642
- Beeman NE, Baumgartner HK, Webb PG, Schaack JB, Neville MC. Disruption of occludin function in polarized epithelial cells activates the extrinsic pathway of apoptosis leading to cell extrusion without loss of transepithelial resistance. BMC Cell Biol 2009; 10:85; PMID:20003227; http://dx.doi.org/10.1186/1471-2121-10-85
- Bjarnason I, Hayllar J, MacPherson AJ and Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993; 104(6):1832-47; PMID:8500743
- Carrozzino F, Pugnale P, Feraille E, Montesano R. Inhibition of basal p38 or JNK activity enhances epithelial barrier function through differential modulation of claudin expression. Am J Physiol Cell Physiol 2009; 297(3):C775-87; PMID:19605737; http://dx.doi.org/10.1152/ajpcell.00084.2009
- Chen Y, Lu Q, Schneeberger EE, Goodenough DA. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol Biol Cell 2000; 11(3):849-62; PMID:10712504; http://dx.doi.org/10.1091/mbc.11.3.849
- Feldman G, Kiely B, Martin N, Ryan G, McMorrow T, Ryan MP. Role for TGF-beta in cyclosporine-induced modulation of renal epithelial barrier function. J Am Soc Nephrol 2007; 18(6):1662-71; PMID:17460148; http://dx.doi.org/10.1681/ASN.2006050527
- Fiorucci S, Antonelli E, Santucci L, Morelli O, Miglietti M, Federici B, Mannucci R, Del Soldato P, Morelli A. Gastrointestinal safety of nitric oxide-derived aspirin is related to inhibition of ICE-like cysteine proteases in rats. Gastroenterology 1999; 116(5):1089-106; PMID:10220501; http://dx.doi.org/10.1016/S0016-5085(99)70012-0
- Guillemot L, Citi S. Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol Biol Cell 2006; 17(8):3569-77; PMID:16723500; http://dx.doi.org/10.1091/mbc.E06-02-0122
- Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 1996; 52(2):237-45; PMID:8694848; http://dx.doi.org/10.1016/0006-2952(96)00181-5
- Hawkey CJ. Non-steroidal anti-inflammatory drugs and peptic ulcers. BMJ 1990; 300(6720):278-84; PMID:2106956; http://dx.doi.org/10.1136/bmj.300.6720.278
- Hoshino T, Tsutsumi S, Tomisato W, Hwang HJ, Tsuchiya T, Mizushima T. Prostaglandin E2 protects gastric mucosal cells from apoptosis via EP2 and EP4 receptor activation. J Biol Chem 2003; 278(15):12752-8; PMID:12556459; http://dx.doi.org/10.1074/jbc.M212097200
- Howe KL, Reardon C, Wang A, Nazli A, McKay DM. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol 2005; 167(6):1587-97; PMID:16314472; http://dx.doi.org/10.1016/S0002-9440(10)61243-6
- Jiang GL, Im WB, Donde Y, Wheeler LA. EP4 agonist alleviates indomethacin-induced gastric lesions and promotes chronic gastric ulcer healing. World J Gastroenterol 2009; 15(41):5149-56; PMID:19891013; http://dx.doi.org/10.3748/wjg.15.5149
- Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H(2)O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol 2000; 279(1):C21-30; PMID:10898713
- Lipschutz JH, Li S, Arisco A, Balkovetz DF. Extracellular signal-regulated kinases 1/2 control claudin-2 expression in Madin-Darby canine kidney strain I and II cells. J Biol Chem 2005; 280(5):3780-8; PMID:15569684; http://dx.doi.org/10.1074/jbc.M408122200
- Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol 2003; 4(3):225-36; PMID:12612641; http://dx.doi.org/10.1038/nrm1055
- Nighot PK, Blikslager AT. Chloride channel ClC-2 modulates tight junction barrier function via intracellular trafficking of occludin. Am J Physiol Cell Physiol 2012; 302(1):C178-87; PMID:21956164; http://dx.doi.org/10.1152/ajpcell.00072.2011
- Nighot PK, Blikslager AT. ClC-2 regulates mucosal barrier function associated with structural changes to the villus and epithelial tight junction. Am J Physiol Gastrointest Liver Physiol 2010; 299(2):G449-56; PMID:20489043; http://dx.doi.org/10.1152/ajpgi.00520.2009
- Olugemo K, Solorio D, Sheridan C, Young CL. Pharmacokinetics and safety of low-dose submicron indomethacin 20 and 40 mg compared with indomethacin 50 mg. Postgrad Med 2015; 127:223-31; PMID:25639879; http://dx.doi.org/10.1080/00325481.2015.1000231
- Oshima T, Miwa H, Joh T. Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am J Physiol Cell Physiol 2008; 295(3):C800-6; PMID:18667601; http://dx.doi.org/10.1152/ajpcell.00157.2008
- Ou YC, Yang CR, Cheng CL, Raung SL, Hung YY, Chen CJ. Indomethacin induces apoptosis in 786-O renal cell carcinoma cells by activating mitogen-activated protein kinases and AKT. Eur J Pharmacol 2007; 563(1–3):49-60; PMID:17341418; http://dx.doi.org/10.1016/j.ejphar.2007.01.071
- Pai VP, Horseman ND. Biphasic regulation of mammary epithelial resistance by serotonin through activation of multiple pathways. J Biol Chem 2008; 283(45):30901-10; PMID:18782769; http://dx.doi.org/10.1074/jbc.M802476200
- Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, Bogert C, Guillen JM, Brendel K, Gross PH, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res 1997; 57(14):2909-15; PMID:9230200
- Reddy BS, Kawamori T, Lubet RA, Steele VE, Kelloff GJ, Rao CV. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res 1999; 59(14):3387-91; PMID:10416599
- Samak G, Suzuki T, Bhargava A, Rao RK. c-Jun NH2-terminal kinase-2 mediates osmotic stress-induced tight junction disruption in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 2010; 299(3):G572-84; PMID:20595622; http://dx.doi.org/10.1152/ajpgi.00265.2010
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286: 6: C1213-28, 2004; PMID:15151915; http://dx.doi.org/10.1152/ajpcell.00558.2003
- Seth A, Yan F, Polk DB, Rao RK. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 2008; 294(4):G1060-9; PMID:18292183; http://dx.doi.org/10.1152/ajpgi.00202.2007
- Singh AB, Harris RC. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem 2004; 279(5):3543-52; PMID:14593119; http://dx.doi.org/10.1074/jbc.M308682200
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141(6):539-45; PMID:7900721
- Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol 2010; 20(3):142-9; PMID:20061152; http://dx.doi.org/10.1016/j.tcb.2009.12.002
- Suemasu S, Tanaka K, Namba T, Ishihara T, Katsu T, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai Aet al. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J Biol Chem 2009; 284(29):19705-15; PMID:19439408; http://dx.doi.org/10.1074/jbc.M109.006817
- Takeuchi K, Ueshima K, Hironaka Y, Fujioka Y, Matsumoto J, Okabe S. Oxygen free radicals and lipid peroxidation in the pathogenesis of gastric mucosal lesions induced by indomethacin in rats. Relation to gastric hypermotility. Digestion 1991; 49(3):175-84; PMID:1769433; http://dx.doi.org/10.1159/000200718
- Tomisato W, Tsutsumi S, Rokutan K, Tsuchiya T, Mizushima T. NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am J Physiol Gastrointest Liver Physiol 2001; 281(4):G1092-100; PMID:11557530
- Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol 1990; 259(3 Pt 1):G462-7; PMID:2169206
- Whittle BJ. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology 1981; 80(1):94-98; PMID:6778761
- Wroblewski LE, Shen L, Ogden S, Romero-Gallo J, Lapierre LA, Israel DA, Turner JR, Peek RM, Jr. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 2009; 136(1):236-46; PMID:18996125; http://dx.doi.org/10.1053/j.gastro.2008.10.011
- Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Lynch RD, Schneeberger EE. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol 2005; 288(6):C1231-41; PMID:15689410; http://dx.doi.org/10.1152/ajpcell.00581.2004
- Zhang X, Morham SG, Langenbach R, Young DA. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J Exp Med 1999; 190(4):451-9; PMID:10449516; http://dx.doi.org/10.1084/jem.190.4.451