
ABSTRACT
Accumulation of senescent cells in cerebrovasculature is thought to play an important role in age-related disruption of blood–brain barrier (BBB). Using an in vitro model of human BBB, composed of brain microvascular endothelial cells (BMECs), astrocytes and pericytes, this study explored the so-called correlative link between BMEC senescence and the BBB dysfunction in the absence or presence of functionally distinct senotherapeutics. Replicative senescence was deemed present at passage ≥19 where BMECs displayed shortened telomere length, reduced proliferative and tubulogenic potentials and increased NADPH oxidase activity, superoxide anion production (markers of oxidative stress), S-β-galactosidase activity and γ-H2AX staining. Significant impairments observed in integrity and function of a model of BBB established with senescent BMECs, ascertained successively by decreases in transendothelial electrical resistance and increases in paracellular flux, revealed a close correlation between endothelial cell senescence and BBB dysfunction. Disruptions in the localization or expression of tight junction proteins, zonula occludens-1, occludin, and claudin-5 in senescent BMECs somewhat explained this dysfunction. Indeed, treatment of relatively old BMEC (passage 16) with a cocktail of senolytics (dasatinib and quercetin) or senomorphics targeting transcription factor NF-κB (QNZ), p38MAPK signaling pathway (BIRB-796) or pro-oxidant enzyme NADPH oxidase (VAS2870) until passage 20 rendered these cells more resistant to senescence and totally preserved BBB characteristics by restoring subcellular localization and expression of tight junction proteins. In conclusion, attempts that effectively mitigate accumulation of senescent endothelial cells in cerebrovasculature may prevent age-related BBB dysfunction and may be of prophylactic or therapeutic value to extend lifelong health and wellbeing.
Introduction
Aging is a physiological process that begins during young adulthood and affects the structure and function of vascular system over time.Citation1,Citation2 Endothelial dysfunction (ED) is widely regarded as the main phenotype that promotes age-dependent changes in vasculature.Citation3,Citation4 Endothelium covers the entire inner surface of all blood vessels and is formed by a monolayer of endothelial cells (ECs). In addition to regulating vascular tone, vascular permeability, angiogenesis, coagulation and inflammation through synthesis and release of several vasoactive compounds, ECs also help establish specific barriers, conducting specific tasks, in different organs.Citation5,Citation6 Blood−brain barrier (BBB) represents one such barrier in the central nervous system and controls the selective passage of compounds between brain parenchyma and the systemic circulation.Citation7 Although astrocytes, pericytes, and microglial cells also contribute to the formation of BBB, brain microvascular ECs (BMECs), capable of forming tight junctions with the adjacent BMECs to prevent paracellular leakage, are regarded as the main cellular component of this unique barrier.Citation6 Tight junctions are multiprotein junctional complexes that consist of several transmembrane proteins and tight junction-associated scaffolding proteins, notably occludin, claudin-5, and zonula occludens-1 (ZO-1).Citation8 So, any biological scenario capable of affecting viability or function of BMECs during aging process may trigger ED as an early event and eventually compromise BBB. Hence, identification of mechanisms involved in the initiation or progression of ED is of crucial importance to develop novel strategies that can delay or prevent BBB dysfunction and improve lifelong health and wellbeing.
Accumulation of senescent ECs in vasculature is increasingly recognized as an important stimulus for the development of ED during chronological aging.Citation9,Citation10 Indeed, EC senescence is implicated in both the structural and functional deterioration of the endothelium.Citation11,Citation12 Consistent detection of senescent ECs, defined by permanent cell cycle arrest, shortened telomere length and positive staining for senescence-associated-β-galactosidase (SA-β-gal), in atherosclerotic plaques attest the intrinsic role played by EC senescence in the formation and progression of vascular pathologies.Citation13,Citation14 Oxidative stress, defined by the excessive availability of superoxide anion (O2−) and other reactive oxygen species (ROS), is thought to be a key factor for EC senescence during chronological aging and its cell culture model, termed replicative senescence.Citation15,Citation16 NADPH oxidase constitutes the main enzymatic source of oxidative stress in vasculature.Citation17 Detection of senescent cells in human coronary arteries but not in internal mammary arteries, known to possess very high antioxidant capacity, confirms oxidative stress as an important inducer of EC senescence and ensuing ED.Citation18 Restoration of tubulogenic and BBB-forming capacities of endothelial progenitor cells (EPCs), coupled with a considerable delay of senescence, by an NADPH oxidase inhibitor (VAS2870) further verify the role of this oxidase in cellular senescence and cerebral barrier dysfunction.Citation19
Increasing evidence suggest that acquisition of an irreversible senescence-associated secretory phenotype (SASP), accompanied by secretion of several cytokines, chemokines, and matrix metalloproteases may help spread senescence to the neighboring cells.Citation20 Here, activation of p38MAPK/NF-κB pathway appears to be the main stimulus.Citation21,Citation22 Indeed, selective inhibition of p38MAPK activity has been shown to collapse the senescence-associated cytokine network in human fibroblasts and to delay replicative senescence induced by dysfunctional telomeres.Citation22,Citation23 Due to its ability to activate both the p53 and pRb/p16 growth arrest pathways, p38MAPK has also been shown to play an important role in the senescence growth arrest.Citation23 Similarly, specific targeting of NF-κB, a transcription factor that regulates the release of inflammatory cytokines and ROS, has been shown to improve endothelial function in healthy older adults to the levels observed in young controls.Citation24 Suppression of cellular proliferation rate, SA-β-gal activity and the expression of senescence-associated proteins, namely p16, p21, and p53, in mouse aortic ECs subjected to inflammatory cytokine interleukin-17A by an NF-κB inhibitor further confirmed the role of NF-κB in EC senescence.Citation25
In addition to targeting of so-called specific mechanisms involved in cellular senescence, elimination of senescent cells by senolytics may also attenuate or even eradicate the impact of senescent cells on both the neighboring cells and the remote tissue. The combination of dasatinib and quercetin (D + Q) has been the most commonly used senolytics in recent experimental and clinical studies.Citation26–28 While displaying no significant effects on non-senescent cells, D + Q selectively eliminated senescent cells in skin and adipose tissues.Citation28 Studies specifically focusing on ECs have shown that D + Q alleviates LPS-induced senescence in human umbilical vein ECs through inactivation of a pathway involving MAPK-NF-κB axis.Citation29 However, the mechanism of D + Q in replicative EC senescence-related BBB dysfunction remains unexplored.
In light of the above, using a cell culture model of chronological aging, the current study has explored the impact of senescence on the morphological and functional characteristics of BMEC in the absence or presence of D + Q or senomorphics targeting p38MAPK, NF-κB and NADPH oxidase. Using a well-established in vitro model of human BBB,Citation11,Citation30 it then investigated the specific mechanisms through which D + Q and p38MAPK inhibition exert their so-called cerebral barrier-protective effects. Taken together the data generated provide important novel insights into the prophylactic and therapeutic role of senotherapeutics in controlling the EC senescence-mediated BBB dysfunction and associated vascular complications.
Materials and methods
Cell culture
HBMECs were purchased from Neuromics (Minneapolis, MN, USA). Human pericytes and astrocytes were purchased from TCS CellWorks Ltd. (Buckingham, UK). All cells were cultured in a humidified atmosphere (75% N2, 20% O2, 5% CO2) at 37°C in their respective media (Sciencell Research Laboratories, San Diego, USA).
Throughout the study, cells between passages 3 and 5 were used as young or non-senescent cells. Senescence was induced through repetitive culture of young cells and deemed to exist when about 70% of the cultured cells were stained positive for SA-β-Gal and γH2AX. In most experiments, this characteristic appeared between passages 19 and 21. The modulatory impact of senolytics (D + Q, 20 nM and 5 μM, respectively) and senomorphics targeting p38MAPK (BIRB-796, 5 μM), NADPH oxidase (VAS2870, 5 μM) or NF-κB (QNZ, 1 μM) on markers of senescence, BBB function, etc were assessed by treating passage 16 cells till passage 20 with the specific agents indicated in brackets. To ensure about the fresh supply of inhibitors, culture media were changed every other day in these experiments.
Senescence-associated β-galactosidase activity
HBMECs were seeded in 12-well plates and subjected to the outlined experimental conditions. The SA-β-gal activity were evaluated using a beta-galactosidase staining kit (APExBIO, USA) as per the manufacturer’s instructions. In brief, HBMECs were washed with warm PBS and fixed with the fixation solution at room temperature for 10 min. The cells were then washed and incubated in staining solution mix overnight at 37°C. The cells were viewed under a light microscope and the cells with blue color were regarded as senescent. The number of cells stained positive or otherwise for SA-β-gal was counted manually in five randomly chosen areas on each slide and the percentage of SA-β-gal positive cells were calculated considering all the readings from three independent experiments
Establishment of triple cell culture model of human BBB
Triple cell culture model of human BBB, composed of HBMECs, astrocytes and pericytes, was used throughout the study due to well-established crosstalk amongst different cell layers and thus its close functional resemblance to BBB in vivo.Citation17,Citation30 To this end, approximately 1 × 105 HAs were seeded on the basolateral side of the transwell inserts (polyester membrane, 12 mm diameter, 0.4 μm pore size, Corning, UK). Once the cells adhered to the inserts, the inserts were reverted to the original orientation and cultured in a fresh 12-well plate to 90% confluence. 5 × 104 young or 10 × 104 senescent HBMECs were then seeded onto the apical side of the inserts containing astrocytes. Since senescent cells lose their capacity to proliferate, it was essential to seed a significantly higher number of senescent HBMECs in these studies to promote appropriate monolayer formation. Once, full confluence is attained in both layers, the inserts were transferred to fresh 12-well plates containing confluent pericytes, to set up the triple cell culture model of human BBB.
Assessment of the BBB integrity and function
The integrity and function of the BBB were evaluated successively by measurements of the transendothelial electrical resistance (TEER) and the paracellular flux of sodium fluorescein (NaF, 376 Da) or Evan’s blue-labeled albumin (EBA, 67 kDa). TEER was measured using a EVOM resistance meter and STX electrodes (World Precision Instruments, Hertfordshire, UK). After reading the TEER values, the inserts were rinsed with Hank’s Balanced Salt Solution (HBSS, Sigma) and transferred into fresh 12-well plates containing 2 ml of HBSS. 500 μl of 50 μg/ml NaF or 165 μg/ml EBA was added in the luminal chamber of each insert and the plates with the inserts were incubated for 1 hour. After this, 400 μl of solutions from the luminal and abluminal chambers were collected and 100 μl of each solution was added into a 96-well plate in triplicate. Both NaF and EBA levels were measured using a FLUOstar Omega microplate reader (BMG Labtech Ltd, UK) and the flux across the barrier was calculated as before.Citation31
Measurement of NADPH oxidase capacity and superoxide anion level
NADPH oxidase activity and superoxide anion production were assessed through lucigenin chemiluminescence and cytochrome C reduction assays, respectively as previously described.Citation30 Briefly, for the chemiluminescence assay, approximately 50 μg of BMEC homogenates were incubated at 37°C in assay buffer containing potassium phosphate buffer (300 mM, pH 7.0, Sigma), ethylene glycol tetraacetic acid (50 mM, Sigma), sucrose (1 M, Sigma) and lucigenin (200 μM, Sigma). The contributions of nitric oxide synthase (NG-nitro-L-arginine methyl ester, 10 mM, Sigma), mitochondrial respiratory chain complex 1 (rotenone, 10 mM, Sigma), xanthine oxidase (allopurinol, 10 mM, Sigma) and cyclooxygenase (indomethacin, 10 mM, Sigma) to overall O2− production was eliminated by adding the specific inhibitors in brackets to assay buffer. Following ~15 min of incubation with these inhibitors, NADPH (100 μmol/L, Calbiochem, UK) was added to start the reaction which was monitored every minute for 2 hours and the rate of reaction was calculated using a luminometer (FLUOstar Omega, BMG Labtech, Aylesbury, UK).
For the cytochrome-C reduction assay, approximately 100 μg of homogenates were incubated for 1 h at 37°C with assay buffer containing HEPES (1 M, Calbiochem, UK), ethylene glycol tetraacetic acid (50 mM, Sigma), sucrose (1 M, Sigma), mannitol (1 M, Sigma) and cytochrome C (800 μM, Sigma). O2− production was then studied as the reduction of cytochrome C and monitored with the change in absorbance at 550 nm using a plate reader (FLUOstar Omega, BMG Labtech, Aylesbury, UK).
Proliferation assay
The proliferative capacity of HBMECs was evaluated by using the WST-1 (4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2 H-5-tetrazolio]-1.3-benzene disulfonate) kit (Abcam, UK). In principle, the kit is used to quantify the cleavage of tetrazolium salt WST-1 by mitochondrial dehydrogenases in viable cells. So, the higher the absorbance readings are, the larger the numbers of viable cells are. In brief, 5 × 103 HBMECs were seeded in 96-well plates and cultured for 24 h in a humidified atmosphere, as described above. The cells were then subjected to experimental conditions, and the media were subsequently replaced with 100 μL fresh medium containing 10 μL of WST-1. The plates were incubated for 2 h at 37°C prior to reading the absorbance (480 nm) using a FLUOstar Omega microplate reader (BMG Labtech Ltd, UK).
Tube formation assay
Tube formation assay was performed to evaluate the angiogenic capacity of the cells. For this, HBMECs were seeded in 96-well plates pre-coated with Matrigel (Corning, US) and incubated at 37°C for 4 hours. The total number and length of tubule network, calculated as the sum of number or length of segments and branches detected in the analyzed area, were assessed using Angiogenesis Analyzer plugin ImageJ software (version 1.52k, NIH, Maryland, USA). Images, acquired from 5 randomly selected fields of each well, were analyzed under a light microscope.
Immunocytochemistry
To detect the localization of tight junction proteins, occludin, claudin-5, and ZO-1, to examine the level of DNA damage marker, γ-H2AX and to evaluate the level of stress fiber formation, HBMECs (9 × 104) were seeded on glass coverslips and cultured to about 90% confluence in 12-well plates. The cells were then fixed and permeabilised with 4% paraformaldehyde for 20 minutes and 0.1% Triton X-100 for 5 minutes before incubating overnight at 4°C with primary antibodies specific for tight junction proteins (1:200, all from ThermoFisher, UK) or γ-H2AX (1:250, Abcam, UK). On the following day, the coverslips were washed and incubated with relevant secondary antibodies for 1 h at room temperature. Nuclei were then detected in these cells by DAPI (4,6-diamidino-2-phenylindole) staining. To detect stress fibers, HBMECs were stained with rhodamine phalloidin (Abcam, UK) for 60 minutes at room temperature. The coverslips were then mounted on glass slides using mounting medium (Vector Laboratories, Peterborough, UK) and visualized by fluorescence microscopy (Zeiss Axio Observer, Carl Zeiss Ltd, Cambridge, UK). ImageJ software (version 1.52k, NIH, Maryland, USA) was used to quantify the florescence signal. The data were calculated as before and presented as corrected total cell fluorescence.Citation17 In brief, the junctional areas between cells were selected as the regions of interest (ROI) to measure the intensity of the fluorescent signal and the total fluorescent signals in each image were divided by the area of ROI to acquire the average fluorescent density of the tight junction protein for each image. The number of nuclei stained positive or otherwise for γ-H2AX was counted manually in five randomly chosen areas on each slide and the percentage of γ-H2AX-positive cells were calculated considering all the readings from three independent experiments. The number of stress fibers traversing the cells were manually counted on five randomly selected areas on each slide and the number of stress fibers were comparatively analyzed considering data gathered from three independent experiments.
Quantitative PCR
Genomic DNA from HBMECs were extracted with the Nucleon BACC1 Genomic DNA Extraction Kit (GE Healthcare, UK). Telomere length of the genomic DNA samples was measured by real-time PCR using the Absolute Human Telomere Length Quantification qPCR Assay Kit (ScienCell Research Laboratories, USA). The average telomere length on each chromosome end was calculated as per the manufacturer’s instructions using the formulae based on the Cq value acquired by Agilent Mx3000P QPCR System (Santa Clara, USA).
Statistical analysis
Data are displayed as mean ± standard error of the mean (SEM) from a minimum of three independent experiments. Statistical analyses were performed by unpaired t-test. GraphPad Prism 9.0 statistical software package (GraphPad Software Inc., La Jolla, Ca, USA) and SPSS Statistics 27.0 (IBM, New York, USA) were used to perform these analyses. P < .05 was considered as significant.
Results
Late-passage endothelial cells exhibit typical characteristics of cellular senescence
Given the non-specificity of markers used to detect senescence in in vitro settings, in the present study, a series of markers targeting cellular morphology, cell cycle progression and metabolic activity were simultaneously studied. Initial studies looking at the changes in endothelial phenotype showed that BMECs subjected to repetitive cell culture acquired larger and flattened morphology and developed thick actin stress fibers traversing the cells at passage ≥19. In this context, relevant studies exploring the effects of BIRB-796, QNZ and VAS2870 or D + Q on phenotypic changes showed that treatments with these agents, except for QNZ, even at a reasonably late stage (from passage 16 (p16) to p20) lead to deceleration of morphological changes and stress fiber formation ()).
Figure 1. Exposure of human BMECs to an in vitro model of chronological aging, mimicked by replicative senescence, induces profound alterations in cellular morphology (a) and cytoskeletal organization compared to young cells (b). Compared to young cells (p5), relatively old (p16) and senescent cells (p20) possess larger and flattened morphology and thick actin stress fibers. Treatment of p16 cells with inhibitors for p38MAPK (BIRB-796), NF-κB (QNZ), NADPH oxidase (VAS2870) or a cocktail of senolytics (dasatinib and quercetin; D + Q) until p20 attenuated the effect of replicative senescence on both morphology (a) and cytoskeletal organization as ascertained by reductions in number of stress fibers indicated by white arrows (b, c). Scale bars: 100 μm. The images of cellular morphology and stress fibers were captured using x20 and x40 magnification. Data are expressed as mean ± SEM from three independent experiments. *P < .05 compared to p5 BMECs. # P < .05 compared to p20 BMECs.
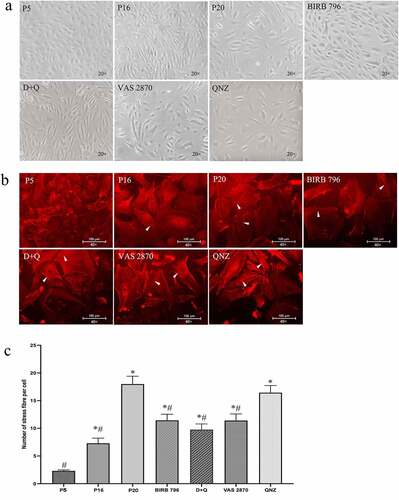
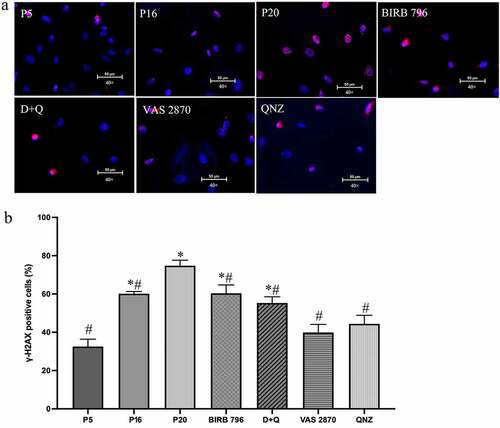
Subsequent studies aiming to provide further evidence for BMEC senescence demonstrated significantly higher SA-β-gal activity in late-passage (p20) versus low-passage (p5) and mid-passage (p16) cells ()). However, as other conditions, e.g. serum starvation may also induce an increase in SA-β-gal activity,Citation32 the level of γ-H2AX, a sensitive marker for double-stranded DNA breaks, was also investigated in this study. As with the SA-β-gal activity, the γ-H2AX staining was also significantly higher in p20 BMECs compared to younger counterparts ()). Similar to morphological studies, treatments with all agents, other than QNZ, drastically reduced the number of SA-β-gal-positive cells compared to senescent group. Even so, SA-β-gal activity remained well above the levels observed in p5 and p16 cells. Interestingly, while all agents significantly decreased γ-H2AX staining, the maximal effect was obtained with VAS2870 and QNZ ().
Figure 2. The number SA-β-gal positive cells increases by replicative senescence. Exposure of human BMECs to repetitive cell culture markedly increased the number of SA-β-gal-positive cells in late passage (p20) cells compared to young (p5) and mid-passage (p16) cells. Treatment of p16 cells with senomorphics targeting p38MAPK (BIRB-796) or NADPH oxidase (VAS2870) or a cocktail of senolytics (dasatinib and quercetin; D + Q) until p20 diminished the number of cells that underwent senescence. Treatment with QNZ, an NF-κB inhibitor significantly elevated the number of SA-β-gal-positive cells (a, b). Data are expressed as mean ± SEM from three independent experiments. The SA-β-gal stainings were visualized using 20x magnification. *P < .05 compared to p5 BMECs. #P < .05 compared to p20 BMECs.
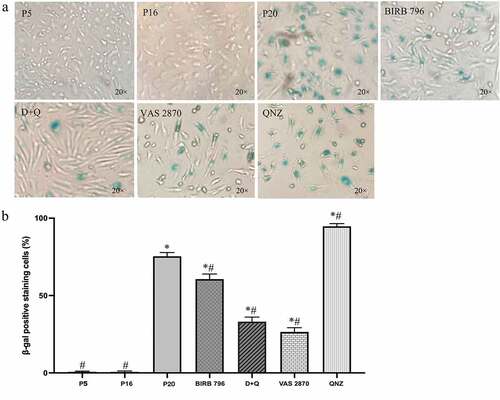
Figure 3. Treatments with senotherapeutics diminish senescence-induced DNA damage. Replicative senescence, realized by repetitive cell culture, markedly increased the percentage of γ-H2AX-positive cells in late passage (p20) human BMECs compared to young (p5) and mid-passage (p16) cells. Treatments of p16 cells with senomorphics targeting p38MAPK (BIRB 796), NF-κB (QNZ) or NADPH oxidase (VAS2870) or senolytics (dasatinib and quercetin; D + Q) until p20 diminished the percentage of γ-H2AX-positive cells (a, b). Scale bars: 50 μm. The γ-H2AX stainings were visualized using 40x magnification. Data are expressed as mean ± SEM from three independent experiments. *P < .05 compared to p5 BMECs. # P < .05 compared to p20 BMECs.
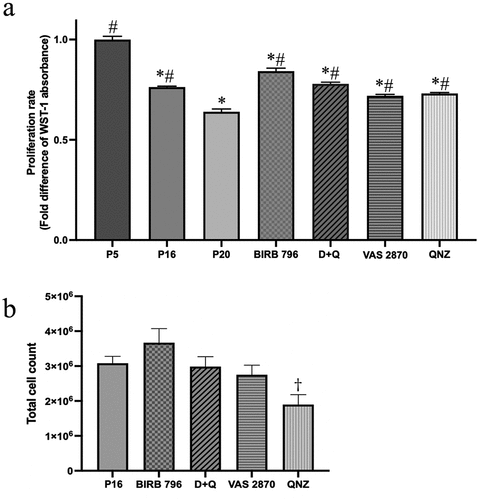
Scrutiny of cell proliferation as an index of cell cycle progression revealed a significant decline in BMEC proliferative capacity with increasing passage numbers. Despite halting these decreases, treatments with senolytics and senomorphics failed to return the proliferation rates to the levels seen in low-passage cells ()). These studies also showed that treatments with QNZ significantly diminished the number of viable BMECs compared to controls, i.e. p16 cells ()). Because of this potential apoptotic effect, we did not pursue QNZ in the subsequent experiments. Moreover, due to a recent study illustrating that suppression of oxidative stress with vitamin C or VAS2870 delays EPC senescence and augments their functional capacity, NADPH oxidase was also not targeted in the following functional studies.Citation19
Figure 4. Senotherapeutics reduce the impact of senescence on BMEC proliferative capacity. The proliferation rate of BMECs, assessed by WST-1 assay, decreased with increasing passage numbers. Treatments of p16 cells with senomorphics targeting p38MAPK (BIRB 796), NF-κB (QNZ) or NADPH oxidase (VAS2870) or senolytics (dasatinib and quercetin; D + Q) until p20 significantly decreased the suppressive effect of senescence on proliferation (a). However, treatments with QNZ significantly reduced the number of viable cells compared to p16 cells, control group (b). Data are expressed as mean ± SEM from three independent experiments. *P < .05 compared to p5 BMECs. # P < .05 compared to p20 BMECs. †P < .05 compared to p16 BMECs.
From this point on, BMECs with high proliferative capacity and less than 3% SA-β-gal positive staining were defined as young cells. These included cells up to and including p6. HBMECs that showed no sign of proliferation for a week in culture with complete medium and had over 70% SA-β-gal- and γ-H2AX-positive staining were defined as senescent. These included cells at and above p19.
Considering that telomeres play an essential role in preserving the integrity of chromosomes during the cell division cycle and undergo programmed shortening in culture conditions,Citation33 the telomere length was investigated by quantitative PCR and shown to be substantially shorter in senescent versus young cells. Inability of D + Q or BIRB-796 to influence telomere length suggested that telomeres were already irretrievably lost in p16 BMECs ()).
Figure 5. Senescence is associated with reduced telomere length and increased oxidative stress. Compared to young cells (p5), the average telomere length is markedly reduced in senescent (p20) cells. Treatments of p16 cells with BIRB-796, a p38MAPK inhibitor and a senomorphic or a cocktail of senolytics (dasatinib and quercetin; D + Q) until p20 failed to negate the effect of senescence on telomere length (a). Increases in NADPH oxidase activity and superoxide anion generation confirm that senescent cells have oxidative stress. Treatment with VAS2870, an inhibitor of NADPH oxidase, neutralize the effect of senescence on both parameters (b, c). Data are expressed as mean ± SEM from three independent experiments. *P < .05 compared to p5 BMECs. # P < .05 compared to p20 BMECs.
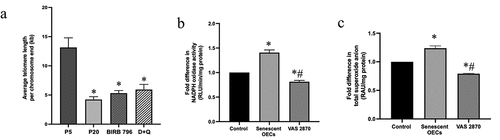
In addition to above, senescent cells also possessed significantly greater levels of NADPH oxidase activity and O2.− production, supporting the notion that oxidative stress plays a pivotal role in the process of cell senescence.Citation15 Marked inhibitions observed in both enzyme activity and O2.− release, the foundation molecule of all ROS,Citation34 by VAS2870 confirmed that senescent cells were metabolically active and responsive to external stimuli ()).
Senescent endothelial cells lose their angiogenic potential and fail to form BBB
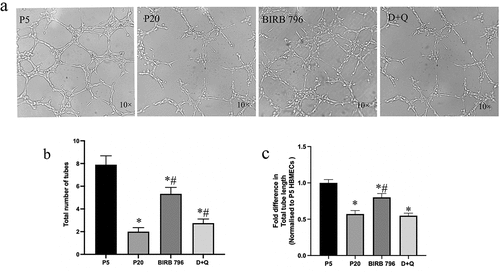
Angiogenesis is a vital process by which new blood vessels form and thus contribute to growth, development and regeneration of the damaged tissue.Citation35 As evidenced by significant decreases in number and length of new tubules appeared on matrigel, senescence negatively affected the capacity of ECs for angiogenesis. Here, suppression of p38MAPK by BIRB-796, rather than elimination of senescent cells via D + Q, appeared to be more effective in diminishing the impact of senescence on tubule formation in in vitro settings ()).
Figure 6. Senescence adversely affect the angiogenic capacity of BMECs. Senescent (p20) BMECs displayed significantly fewer and shorter tubules on matrigel compared to young (p5) cells. Treatment with senomorphic BIRB-796, a p38MAPK inhibitor, increased the number and length of tubules. In contrast, treatment with a combination of senolytics (dasatinib+quercetin, D + Q) only improved the number of tubules without affecting their length (a-c). The tubules were visualized using 10x magnification *P < .05 compared to p5 BMECs. #P < .05 compared to p20 BMECs.
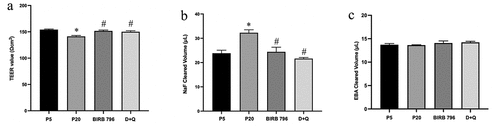
Through lateral migration and consequent replacement of dead or dying ECs, resident ECs play an important role in preventing structural and functional vascular damage.Citation6 The data generated up to this point suggested that senescent BMECs were unlikely to be as protective or functional as their younger counterparts. The overt inadequacy of senescent BMECs to form a functional model of human BBB, evidenced by decreased transendothelial electrical resistance (TEER) and increased paracellular flux of sodium fluorescein (NaF) across the barrier, supported this hypothesis. Interestingly, treatments with BIRB-796 and D + Q completely neutralized the disruptive effects of senescence on cerebral barrier. Contrary to increased permeability of NaF, the flux of Evans blue-labeled albumin (EBA), a high molecular weight permeability marker, remained unchanged across all experimental conditions thereby implying the formation of relatively small inter-endothelial cell openings between senescent BMECs ()).
Figure 7. The presence of senescent BMECs adversely affect blood-brain barrier integrity and function. Co-culture of senescent (p20), but not young (p5), BMECs with astrocytes and pericytes in an in vitro model of human BBB decreased barrier integrity (a) and increased paracellular flux of a low molecular weight (NaF) but not high molecular weight (EBA) permeability marker across the barrier (b, c). Treatments with senomorphic BIRB-796, a p38MAPK inhibitor and a cocktail of senolytics (dasatininb and quercetin, D + Q) neutrailised the deleterious effects of senescence on both BBB integrity and function (a-c). Data are expressed as mean ± SEM from six independent experiments *P < .05 compared to p5 BMECs. #P < .05 compared to p20 BMECs.
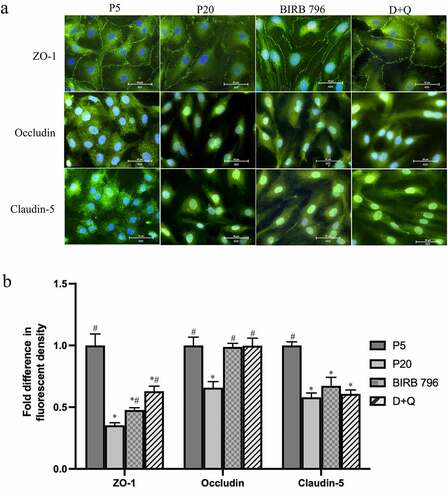
Tight junctions are disrupted in replicative senescent endothelial cells
To determine whether possible changes in tight junctional complex formation might explain the abovementioned BBB-related findings, subcellular localization and abundance of ZO-1, occludin and claudin-5 were investigated. Since senescent cells lost their capacity to proliferate, a significantly higher number of senescent cells were seeded in these studies to promote appropriate monolayer formation. Intermittent interruptions in plasma membrane staining of ZO-1 offered some explanation for the impaired integrity and function of the BBB established with senescent BMECs. Unlike ZO-1, occludin and claudin-5 were found to locate mostly to cytoplasm and nuclei in young and senescent cells, respectively. Although treatments with BIRB-796 and D + Q could somewhat prevent the loss of ZO-1 in cells undergoing senescence, they could not prevent the emergence of abovementioned phenotypic changes, i.e. larger and flattened morphology. Neither BIRB-796 nor D + Q altered the subcellular localization of occludin or claudin-5 observed in senescent cells despite returning the level of the former protein to the levels seen in young cells without affecting that of the latter ()).
Figure 8. Senescence is associated with disruptions in tight junction protein localization and expression. Compared to young (p5) cells, the plasma membrane localization of ZO-1 is interrupted in senescent (p20) human BMECs which was somewhat prevented by treatments with senomorphic BIRB-796, a p38MAPK inhibitor or a cocktail of senolytics (dasatinib+quercetin, D + Q) (a). Unlike ZO-1, both occludin and claudin-5 appear to localize to cytosol and nuclei and treatments with either agent did not greatly influence their subcellular localization (a). Senescence reduced the abundance of all three proteins in BMEC. Treatments with BIRB-796 and D + Q returned the level of occludin to those seen in p5 cells. They somewhat improved the level of ZO-1 but failed to alter that of claudin-5. Data are expressed as mean ± SEM from three independent experiments. Scale bars: 50 μm. All images were captured using 40x magnification. *P < .05 compared to p5 BMECs. #P < .05 compared to p20 BMECs.
Discussion
BBB regulates the passage of an array of selective components between brain parenchyma and the systemic circulation.Citation5–7 Due to its pivotal roles in nutrient supply and waste disposal, it is essential to monitor the function of BBB at all times to conserve neurovascular homeostasis. Increased permeability of the BBB is implicated in chronological aging and can be severely neurotoxic.Citation36,Citation37 Indeed, continual detection of senescent cells at specific sites of age-related pathologies, like atherosclerosis, confirms the idea that cellular senescence contributes to organismal aging and age-related pathologies.Citation12,Citation14 Although formed through complex interactions between neurovascular cells and the underlying basement membrane, dysfunction of BMECs alone may be sufficient to compromise BBB integrity during the aging process.Citation38 Bearing these in mind and using a cell culture model of chronological aging, the current study specifically assessed the impact of BMEC senescence on BBB integrity and function. Relevant studies reporting a strong inverse correlation between donor age and propagative lifespan of skin fibroblasts confirm that replicative cellular senescence occurs during chronological aging and can be reliably used as an experimental model to explore the molecular mechanisms of aging.Citation6,Citation39
Given the non-specificity of commonly used markers for senescence, this study, as indicated before, employed a panel of interrelated markers aiming to detect changes cell phenotype, cell cycle progression and metabolic activity.Citation40 Since accurate identification of specific mechanisms involved in BMEC senescence and dysfunction is critically important for strict regulation of aging-related cerebrovascular alterations and clinical conditions, such as vascular dementia, this comprehensive approach was of paramount importance.Citation41 Acquisition of a flatter and enlarged morphology in cells that ceased to proliferate, in culture media supplemented with all necessary growth factors, confirmed that senescence occurred in BMEC after about 19–20 population doublings. Marked reductions in protein degradation and RNA turnover, leading to increases in intracellular RNA and protein content may, to a degree, account for the cellular enlargements observed in this study. Persistent DNA damage response arising from the exhaustion of DNA replication cycles and telomere shortening due to repetitive cell culture may also contribute to cellular enlargements by activating transcription factor p53 and cyclin-dependent kinase inhibitor p21 and thus blocking the cell cycle at G1-S interphase.Citation42 Further scrutiny of cellular morphology by analyses of cytoskeletal organization through actin microfilaments staining verified that senescent cells took up a larger and flattened phenotype and developed thick actin stress fibers traversing the cells. The cytoskeleton primarily helps cells maintain their shape and internal organization which enables cells to perform essential functions like proliferation and motility. Under normal circumstances, the cortical actin bands participate in formation of tight junctions and prevent paracellular flux. Once formed, thick contractile actin stress bundles pull junctional proteins inward whereby breakup junctional complexes and create intercellular gaps in that activations of small GTP-binding protein RhoA and transcriptional factor NF-κB play a role.Citation43,Citation44 Detection of remarkably higher SA-β-gal staining, NADPH oxidase activity, O2.− production and γ-H2AX deposition coupled with permanent DNA damage in cells manifested morphological changes substantiates the notion that senescence is a complex multifactorial process which affects and is affected by various biological phenomena, e.g. oxidative stress.Citation15,Citation33
Despite availability of nutrients, growth factors and space to grow into, quiescence of senescent BMEC indicates that senescent cells also develop functional abnormalities. These are often associated with survival and cell division. Given that proliferation and migration of BMECs are important prerequisites for replacement of the dead or dying ECs, age-dependent progressive loss of BMECs and their functionality are linked to perturbation of vascular integrity and homeostasis.Citation6 Accumulation of metabolically active senescent BMECs in cerebrovasculature can further worsen the vascular impairment by adversely affecting the function of circulating EPCs through agents, including ROS, cytokines and chemokines that they synthesize and release.Citation15 By differentiating into mature ECs and through their paracrine effect, EPCs are known to assist vascular re-endothelialisation and regeneration.Citation6,Citation11,Citation19
Attenuation of angiogenesis by senescent BMECs may also exacerbate cerebrovascular damage. Observation of fewer and shorter tubule formation on matrigel with senescent BMECs proved that senescence adversely affects angiogenesis possibly through a mechanism involving the inhibition of certain pro-angiogenic factors.Citation45,Citation46 Failure of senescent BMECs to establish a functional model of BBB, when co-cultured with astrocytes and pericytes, provided further evidence for the dysfunctionality of senescent cells. In support of a recent study documenting that mixing replicative senescent EPCs with young BMECs elicits BBB damage in in vitro settingsCitation19 this study also showed a substantial breakdown in integrity and function of BBB established with senescent BMECs and attributed this to disintegration of ZO-1 staining in plasma membrane of senescent cells. Considering that ZO-1 mediates the connections between actin cytoskeleton and other tight junction proteins, these relatively small sporadic disruptions may conceivably have a huge impact on overall BBB function.Citation47 Decreased availability and somewhat unexpected cytoplasmic and nuclear localization of claudin-5 and occludin in senescent BMECs may also contribute to BBB dysfunction. Cytoplasmic and nuclear localization of occludin has previously been noted in other cerebral cells, such as astrocytes and neuroepithelial cells. In these cells, occludin appeared to play crucial roles relating to RNA metabolism and nuclear functions.Citation48 In another study, non-plasma membrane localization of tight junction proteins has been correlated with the decreased telomerase activity in senescent cells.Citation49 While substantially reduced telomere length in senescent BMECs may shed some light on the non-plasma membrane localization of occludin and claudin-5 observed in this study, these findings cannot satisfactorily explain the complete disappearance of these proteins in BMEC plasma membrane.
To assess whether manipulation of senescence may extend the life span and proliferative capacity of ECs, we treated relatively old but non-senescent BMECs (p16) with a cocktail of senolytics, D + Q or the inhibitors of p38MAPK, NF-κB or NADPH oxidase. Due to its role in senescence growth arrest via activation of p53 and pRb/p16 pathways, p38MAPK has previously attracted some attention.Citation23 In the present study, attenuation of senescence-related changes in cellular morphology, proliferative capacity, tubulogenesis, SA-β-gal activity, γ-H2AX staining and complete restoration of BBB function with BIRB-796 proposes p38MAPK signaling pathway as a potential therapeutic target for the management of chronological senescence. Inhibition of p38MAPK may also halt the spread of senescence to the neighboring cells through suppression of SASP via a mechanism involving NF-κB transcriptional activity.Citation20 Hence, treatments with an NF-κB inhibitor, i.e. QNZ rather unsurprisingly diminished the impact of senescence on cell morphology, stress fiber formation and γ-H2AX staining. However, significant drops in number of viable cells in QNZ-treated experimental groups casts serious doubt on the applicability of NF-κB inhibitors as efficacious therapeutics.
Oxidative stress, emerging from an imbalance between the pro- and anti-oxidants, represents the most commonly encountered pathology in senescent cells.Citation15 Although lower (physiological) levels of O2− regulate cell proliferation, migration and gene expression, prolonged exposure to high levels of O2− and other ROS promotes senescent phenotype through damaging all major macromolecules, i.e. DNA, protein and lipid.Citation15,Citation18,Citation35 Naturally, suppression of NADPH oxidase, the main enzymatic source of endothelial O2−, by VAS2870 effectively delayed the appearance of senescent phenotype, improved proliferative potential of p16 BMECs and corroborated the results of a recent study exhibiting delay of EC senescence and BBB dysfunction with VAS2870.Citation19 Restoration of normal endothelial function in dysfunctional arterial segments with NADPH oxidase inhibitors and ROS scavengers further affirm the role of NADPH oxidase, O2− and oxidative stress in endothelial dysfunction.Citation50–52
Selective removal of senescent cells by senolytics is thought to subside the burden of these cells on their microenvironment (neighboring cells) and remote tissue and improve cerebrovascular function as a consequence. The combination of D + Q is regarded as an effective cocktail of senolytics with significant anti-aging effects.Citation53 While dasatinib, a small-molecule tyrosine kinase inhibitor, induces apoptosis and attenuates the viability of senescent adipocytes,Citation28 quercetin displays potent anti-inflammatory, anti-oxidant and immunoprotective effects and therefore alleviates platelet aggregation, oxidative stress, and vascular permeability.Citation54 Once combined, D + Q exerts further beneficial effects, reduces secretion of pro-inflammatory cytokines, prevents the accumulation of macrophages, attenuates the production of major SASP factors and potentiates the proliferative capacity of progenitor cells to reduce the burden of senescent cells in skin and adipose tissues.Citation26,Citation28,Citation55 In accordance with these observations, treatments with D + Q, in the current study, markedly delayed BMEC senescence and improved their proliferative, angiogenic, and barrier-forming capacity.
There are some limitations to this study. As alluded above, despite using a variety of antibodies raised against different epitopes of occludin and claudin-5, we could not visualize these proteins on BMEC plasma membrane. Albeit in full agreement with the manufacturing companies’ immunocytochemistry reports for the respective proteins, the lack of plasma membrane staining was unexpected and deserve further attention in future studies. It is of note here that 23 proteins have thus far been recognized as the members of claudin family in human tissues. Although claudin-1, claudin-12, claudin-25, claudin-34c1 also appear to be involved in the formation of cerebral tight junctions, claudin-5 constitutes the main family member in brain capillary tissues and is closely associated with the formation and functionality of the BBB.Citation56–59 Hence, this particular protein has been targeted in the present study. However, it is possible that investigation of other aforementioned family members may have yielded plasma membrane staining. Given that p38MAPK/NF-κB pathway acts in part through regulation of SASP, it would have been useful to study the impact of BIRB-796 and D + Q on the composition of SASP. In future, it is important to confirm the correlative link between EC senescence and age-related BBB dysfunction in vivo. Similarly, the efficacy of senolytics and senomorphics need to be explored in the future translational studies to substantiate the link between EC senescence and chronological aging. However, given the lack of cell-specificity of senolytics and senomorphics at the present time, relevant future studies must either employ an in vivo model of BMEC dysfunction or utilize agents that are specific for this particular cell line to distinguish the effects of BMEC senescence from those of peripheral senescence.
In conclusion, this experimental study supports the concept that ECs go into senescence during aging process which ultimately damages the integrity of the BBB. The study reports that inhibition of p38MAPK and removal of senescent cells by senolytics successfully delay the senescence of BMEC and fully restore BBB integrity and function. Taken together, these findings pave the way for the development of effective novel strategies that may delay or abate age-related BBB dysfunction and improve lifelong health and wellbeing as a consequence.
Authors contributions
JY performed most of the experiments and carried out the data analysis. RRA performed oxidative stress-related studies. UB designed and supervised the study, interpreted the data and wrote the manuscript. All authors approved the final version for publication.
Data Availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Bruce TB, Gillespie C, Go A, Greenlund K, et al. Heart disease and stroke statistics - 2010 update: a report from the American heart association. Circulation. 2010;121(7):281–297. doi:10.1161/CIRCULATIONAHA.109.192667.
- Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Col Cardiol. 1994;24(2):471–476. doi:10.1016/0735-1097(94)90305-0.
- Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42(7):1149–1160. doi:10.1016/s0735-1097(03)00994-x.
- Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107(1):139–146. doi:10.1161/01.cir.0000048892.83521.58.
- Bayraktutan U. Endothelium, endothelial progenitor cells and stroke. J Neurol Clin Neurosci. 2017;1:21–22.
- Bayraktutan U. Endothelial progenitor cells: potential novel therapeutics for ischaemic stroke. Pharmacol Res. 2019;144:181–191. doi:10.1016/j.phrs.2019.04.017.
- Segarra M, Aburto MR, Acker-Palmer A. Blood-brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 2021;44(5):393–405. doi:10.1016/j.tins.2020.12.002.
- Lochhead JL, Yang J, Ronaldson PT, Davis TP. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front Physiol. 2020;11 article 914. doi:10.3389/fphys.2020.00914.
- Morgan RG, Ives SJ, Lesniewski LA, Noyes RD, Richardson RS, Donato AJ. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305(2):H251–258. doi:10.1152/ajpheart.00197.2013.
- Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, Bianco P, Dallegri F, Pistoia V, Pezzolo A. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PloS one. 2012;7(4):e35312. doi:10.1371/journal.pone.0035312.
- Abdulkadir RR, Alwjwaj M, Othman OA, Rakkar K, Bayraktutan U. Outgrowth endothelial cells form a functional cerebral barrier and restore its integrity after damage. Neural Regen Res. 2020;15(6):1071–1078. doi:10.4103/1673-5374.269029.
- Vasile E, Tomita T, Brown LF, Kocher O, Dvorak HF. Differential expression of thymosin β-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001;15(2):458–466. doi:10.1096/fj.00-0051com.
- Song P, Zhao Q, Zou MH. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev. 2020;60:101072. doi:10.1016/j.arr.2020.101072.
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis. Role of telomere in endothelial dysfunction. Circulation. 2002;105(13):1541–1544. doi:10.1161/01.cir.0000013836.85741.17.
- Höhn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, Kehm R, König J, Grune T, Castro JP. Happily (n)ever after: aging in the context of oxidative stress, proteostasis loss and cellular senescence Redox biology. Redox Biology. 2017;11:482–501. doi:10.1016/j.redox.2016.12.001.
- Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117(11):2417–2426. doi:10.1242/jcs.01097.
- Abdulkadir RR, Alwjwaj M, Bayraktutan U. Treatment with outgrowth endothelial cells protects cerebral barrier against ischemic injury. Cytotherapy. 2022;24(5):489–499. doi:10.1016/j.jcyt.2021.11.005.
- Mahadevan VS, Campbell M, McKeown PP, Bayraktutan U. Internal mammary artery smooth muscle cells resist migration and possess high antioxidant capacity. Cardiovasc Res. 2006;72(1):60–68. doi:10.1016/j.cardiores.2006.06.022.
- Abdulkadir RR, Alwjwaj M, Othman OA, Rakkar K, Sprigg N, Bath PM, Bayraktutan U. Inhibition of oxidative stress delays senescence and augments functional capacity of endothelial progenitor cells. Brain Res. 2022;1787:147925. doi:10.1016/j.brainres.2022.147925.
- da Silva PFL, Ogrodnik M, Kucheryavenko O, Gilbert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, et al. The bystander effect contributes to the accumulation of senescent cells in vivo aging cell. Aging Cell. 2019;18(1):e12848. doi:10.1111/acel.12848.
- Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype. Cell Signal. 2012;24(4):835–845. doi:10.1016/j.cellsig.2011.12.006.
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–1548. doi:10.1038/emboj.2011.69.
- Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8(2):131–144. doi:10.1046/j.1365-2443.2003.00620.x.
- Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear Factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119(9):1284–1292. doi:10.1161/CIRCULATIONAHA.108.804294.
- Zhang L, Liu M, Liu W, Hu C, Li H, Deng J, Cao Q, Wang Y, Hu W, Li Q, et al. Th17/IL-17 induces endothelial cell senescence via activation of NF-κB/p53/Rb signaling pathway. Lab Invest. 2021;101(11):1418–1426. doi:10.1038/s41374-021-00629-y.
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–456. doi:10.1016/j.ebiom.2019.08.069.
- Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA, Shapiro IM, Diekman BO, Risbud MV. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun. 2021;12(1):5213. doi:10.1038/s41467-021-25453-2.
- Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–132. doi:10.1038/s41586-020-2403-9.
- Fan T, Du Y, Zhang M, Zhu AR, Zhang J. Senolytics cocktail dasatinib and quercetin alleviate human umbilical vein endothelial cell senescence via the TRAF6-MAPK-NF-κB axis in a YTHDF2-dependent manner. Gerontology. 2022:1–15. doi:10.1159/000522656.
- Abdulkadir RR, Alwjwaj M, McCarthy Z, Bayraktutan U. Therapeutic hypothermia augments the restorative effects of PKC-β and nox2 inhibition on an in vitro model of human blood-brain barrier. Metab Brain Dis. 2021;36(7):1817–1832. doi:10.1007/s11011-021-00810-8.
- Abdullah Z, Bayraktutan U. NADPH oxidase mediates TNF-α-evoked in vitro brain barrier dysfunction: roles of apoptosis and time. Mol Cell Neurosci. 2014;61:72–84. doi:10.1016/j.mcn.2014.06.002.
- Yang NC, Hu ML. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp Gerontol. 2005;40(10):813–819. doi:10.1016/j.exger.2005.07.011.
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;53(6):1194–1217. doi:10.1016/j.cell.2013.05.039.
- Bayraktutan U. Reactive oxygen species, nitric oxide and hypertensive endothelial dysfunction. Curr Hypertens Rev. 2005;1(3):201–215. doi:10.2174/157340205774574568.
- Simons M. Angiogenesis: where do we satnd? Circulation. 2005;111(12):1556–1566. doi:10.1161/01.CIR.0000159345.00591.8F.
- Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease – systematic review and meta-analysis. Neurobiol Aging. 2009;30(3):337–352. doi:10.1016/j.neurobiolaging.2007.07.015.
- Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. doi:10.1038/s41591-018-0297-y.
- Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta. 2019;1865(7):1802–1809. doi:10.1016/j.bbadis.2018.08.008.
- Cristofalo VJ, Beck J, Allen RG. Commentary: cell senescence: an evaluation of replicative senescence in culture as a model for cell aging in situ. J Gerontology A Biol Sci Med Sci. 2003;58(9):B776–B779. doi:10.1093/gerona/58.9.B776.
- Gonzalez-Gualda E, Baker AG, Fruk L, Munoz-Espin D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288(1):56–80. doi:10.1111/febs.15570.
- Graves SI, Baker DJ. Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic Clin Pharmacol Toxicol. 2020;127(2):102–110. doi:10.1111/bcpt.13403.
- Kim YY, Jee HJ, Um JH, Kim YM, Bae SS, Yun J. Cooperation between p21 and Akt is required for p53-dependent cellular senescence. Aging Cell. 2017;16(5):1094–1103. doi:10.1111/acel.12639.
- Allen C, Srivastava K, Bayraktutan U. Small GTPase RhoA and its effector Rho kinase mediate oxygen glucose deprivation-evoked in vitro cerebral barrier dysfunction Stroke. Stroke. 2010;41(9):2056–2063. doi:10.1161/STROKEAHA.109.574939.
- Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282(4):H1485–H1494. doi:10.1152/ajpheart.00645.2001.
- Chang EI, Loh SA, Ceradini DJ, Chang EI, Lin S, Bastidas N, AArabi S, Chan DA, Freedman ML, Giaccia AJ, et al. Age decreases endothelial progenitor cell recruitment through decreases in hypoxia-inducible factor 1α stabilization during ischemia. Circulation. 2007;116(24):2818–2829. doi:10.1161/CIRCULATIONAHA.107.715847.
- Xia WH, Yang Z, Xu SY, Chen L, Zhang XY, Li J, Liu X, Qiu YX, Shuai XT, Tao J. Age-related decline in reendothelialization capacity of human endothelial progenitor cells is restored by shear stress hypertension. Hypertension (Dallas, Tex.: 1979). 2012;59(6):1225–1231. doi:10.1161/HYPERTENSIONAHA.111.179820.
- Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998 6;273(45):29745–29753. doi:10.1074/jbc.273.45.29745.
- Morgan SV, Garwood CJ, Jennings L, Simpson JE, Castelli LM, Heath PR, Mihaylov SR, Vaquéz-Villaseñor I, Minshull TC, Ince PG, et al. Proteomic and cellular localisation studies suggest non-tight junction cytoplasmic and nuclear roles for occludin in astrocytes. Eur J Neurosci. 2018;47(12):1444–1456. doi:10.1111/ejn.13933.
- Huang W, Rha GB, Chen L, Seelbach MJ, Zhang B, Andras IE, Bruemmer D, Hennig B, Toborek M. Inhibition of telomerase activity alters tight junction protein expression and induces transendothelial migration of HIV-1-infected cells. Am J Physiol Heart Circ Physiol. 2010;298(4):H1136–H1145. doi:10.1152/ajpheart.01126.2009.
- Sharif AM, Bayraktutan U, Young IS, Soong VC. N-acetylcysteine does not improve the endothelial and smooth muscle function in the human saphenous vein. Vasc Endovasc Surg. 2007;41(3):239–245. doi:10.1177/1538574407299618.
- Muir AD, McKeown PP, Bayraktutan U. The role of oxidative stress in modulation of the relaxant responses of saphenous vein and internal mammary artery. Open Circ Vasc J. 2010;3(1):59–66. doi:10.2174/1877382601003010059.
- Muir AD, McKeown PP, Bayraktutan U. Role of gender, smoking profile, hypertension, and diabetes on saphenous vein and internal mammary artery endothelial relaxation in patients with coronary artery bypass grafting. Oxid Med Cell Longev. 2010;3(3):199–205. doi:10.4161/oxim.3.3.11757.
- Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020;288(5):518–536. doi:10.1111/joim.13141.
- Qi J, Chen YH, Wang Y, Chen X, Wang L, Hu YJ, Yu BY. Screening of peroxynitrite scavengers in Flos Lonicerae by using two new methods, an HPLC-DAD-CL technique and a peroxynitrite spiking test followed by HPLC-DAD analysis. Phytochem Anal. 2016;27(1):57–63. doi:10.1002/pca.2599.
- Johmura Y, Yamanaka T, Omori S, Wang TW, Sugiura Y, Matsumoto M, Suzuki N, Kumamoto S, Yamaguchi K, Hatakeyama S, et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science. 2021;371(6526):265–270. doi:10.1126/science.abb5916.
- Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16(4):181–188. doi:10.1016/j.tcb.2006.02.006.
- Scalise AA, Kakogiannos N, Zanardi F, Iannelli F, Giannotta M. The blood-brain and gut-vascular barriers: from the perspective of claudins. Tissue Barriers. 2021;9(3):1926190. doi:10.1080/21688370.2021.1926190.
- Berndt P, Winkler L, Cording J, Breitkreuz-Korff O, Rex A, Dithmer S, Rausch V, Blasig R, Richter M, Sporbert A, et al. Tight junction proteins at the blood-brain barrier: far more than claudin-5. Cell Mol Life Sci. 2019;76(10):1987–2002. doi:10.1007/s00018-019-03030-7.
- Greene C, Hanley N, Campbell M. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS. 2019;16(1):3. doi:10.1186/s12987-019-0123-z.