
Abstract
Development of chemical drugs and biologic agents to target human epidermal growth factor receptor (EGFR) has long been established as a promising therapeutic strategy for lung cancer. Previously, it was found that the tumor-suppressor protein MIG6 is a negative regulator of the EGFR kinase, which can bind at the activation interface of asymmetric dimer of EGFR kinase domains to disrupt EGFR dimerization and then inactivate the kinase. The protein adopts two separated segments, that is, MIG6s1 and MIG6s2, to directly interact with EGFR. Here, by computational modeling and analysis of the intermolecular interaction between EGFR kinase domain and MIG6s2 peptide, we demonstrated that the dephosphorylated MIG6s2 peptide can be converted from nonbinder to weak binder and then to moderate binder of EGFR by phosphorylation and cyclization, respectively; the former introduces strong electrostatic potential to EGFR–peptide complex by forming two geometrically satisfactory salt bridges across the complex interface, while the latter minimizes entropy penalty upon the binding of highly flexible peptide to EGFR. Subsequently, the linear phosphorylated and dephosphorylated peptides and phosphorylated cyclic peptide were synthesized and purified, and their binding affinities to the recombinant protein of human EGFR kinase domain were determined by fluorescence polarization titration. As expected theoretically, the linear dephosphorylated peptide has no observable binding to the kinase, and further phosphorylation and cyclization can confer, respectively, low and moderate affinities to the peptide, suggesting a good consistence between the computational analysis and experimental assay.
Introduction
Receptor tyrosine kinase (RTK) superfamily represents a subgroup of transmembrane proteins with an intrinsic tyrosine kinase activity that determines various cellular functions as diverse as growth, differentiation, and cell motility or survival (Ai et al. Citation2014). The human epidermal growth factor receptor (EGFR) as an important member of RTK has received particular attention in recent years owing to its strong association with malignant proliferation, which has been shown to play a central role in the development and progression of nonsmall-cell lung cancer (NSCLC) (Bethune et al. Citation2010). EGFR activation results from the formation of an asymmetric dimer in which the C-terminal lobe of one kinase domain plays a role similar to that of cyclin in activated CDK/cyclin complexes. The CDK/cyclin-like complex formed by two kinase domains, thus explains the activation of EGFR-family receptors by homo- or hetero-dimerization (Zhang et al. Citation2006). EGFR dimerization stimulates its intrinsic intracellular protein–tyrosine kinase activity. As a result, autophosphorylation of several tyrosine residues in the C-terminal domain of EGFR occurs to elicit downstream activation and signaling by several other proteins that associate with the phosphorylated tyrosines through their own phosphotyrosine-binding SH2 domains.
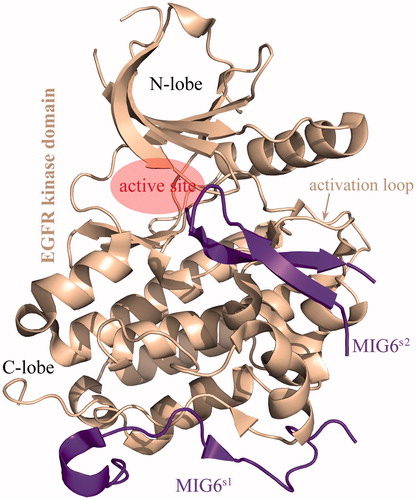
The receptor-associated late transducer MIG6 has been reported as an important negative regulator of the EGFR signaling by binding at the activation interface of EGFR kinase domain to disrupt EGFR dimerization (Hackel et al. Citation2001). Accumulated evidences suggest that the MIG6 plays an essential role in suppressing the development, proliferation, and metastasis of NSCLC by inactivating EGFR (Li et al. Citation2014; Maity et al. Citation2015; Izumchenko and Sidransky Citation2015; Zhang et al. Citation2007). Knockout of the MIG6 gene has also been found to strongly associate with a variety of tumors such as breast cancer (Wendt et al. Citation2015), endometrial cancer (Kim et al. Citation2014) and skin tumor (Ferby et al. Citation2006). The crystal structure of EGFR–MIG6 complex revealed that two separated segments in MIG6 protein, called MIG6s1 (residues 336–361) and MIG6s2 (residues 376–398), interact with the C-lobe of EGFR kinase domain () (Park et al. Citation2015). The binding of MIG6s1 to EGFR activation interface prevents an asymmetric dimer formation, leading to the inhibition of EGFR activation at micromolar-binding affinity level (Moonrin et al. Citation2015). However, the MIG6s1 is only able to inhibit the kinase domain in the context of asymmetric dimer formation, but not directly relevant for shutting down kinase activity. The presence of MIG6s2 C-terminal to MIG6s1 exhibits ability to bind tightly to activated EGFR, which induces the EGFR conformation change from active to inactive states; MIG6s1 alone cannot confer this property, because the kinase residues that interact with it do not change conformation upon activation (Zhang et al. Citation2007). Crystallographic analysis revealed that the MIG6s2 can interact directly with activation loop and the regions close to kinase active site (Park et al. Citation2015). Recently, Yu et al. have successfully performed truncation, modification, and optimization for improving MIG6 affinity to EGFR protein (Yu et al. Citation2016). Here, we attempted to systematically investigate the intermolecular interaction between the EGFR kinase domain and MIG6s2 peptide by integrating in silico modeling and in vitro assay. The isolated MIG6s2 peptide cannot bind effectively to EGFR kinase domain, and we herein proposed a molecular design scheme to rationally convert it from the nonbinder to binder of EGFR by phosphorylation and cyclization. We also demonstrated that the scheme can work fairly well by using fluorescence polarization assay and elucidated the molecular mechanism and biological implication underlying the affinity improvement upon the structural modification and optimization of MIG6s2 peptide.
Figure 1. The complex crystal structure of EGFR kinase domain with MIG6s1 and MIG6s2 (PDB: 4ZJV).
Materials and methods
Molecular simulation
Molecular dynamics (MD) simulations of isolated MIGs2 peptide and EGFR–MIGs2 complex (where the MIGs2 peptide can be phosphorylated, dephosphorylated, linear, and/or cyclized) were carried out using AMBER03 force field (Duan et al. Citation2003) implemented in the AMBER11 package (Case et al. Citation2005). A truncated octahedral box of TIP3P waters (Jorgensen et al. Citation1983) was added with a 10 Å buffer around the complex. Counterions were added to make the simulated system electroneutral. The phosphotyrosine parameters for pTyr394 and pTyr395 of MIGs2 were derived from the AMBER parameter database (Steinbrecher et al. Citation2012). The simulated system was heated from 0 to 300 K over 500 ps, followed by a MD production simulation performed in an isothermal isobaric ensemble with periodic boundary conditions. The time step was set to 2 fs. The particle mesh Ewald (PME) method (Darden et al. Citation1993) was employed to calculate long-range electrostatic interactions. The SHAKE method (Ryckaert et al. Citation1977) was employed to constrain all covalent bonds involving hydrogen atoms.
Binding free energy calculation
The EGFR–MIGs2-binding free energy was calculated using molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method based on 100 snapshots of EGFR–MIGs2 peptide complex structure extracted evenly from MD equilibrium trajectories of the complex (Yang et al. Citation2015, Citation2016): Δ G = Δ Eint + Δ Ddslv - TΔ S, where ΔEint is the intermolecular interaction energy between the EGFR and MIGs2 peptide, which was calculated using force field approach, and ΔDdslv is the desolvation free energy due to peptide binding, which can be computed by numerical solution of the nonlinear Poisson–Boltzmann equation (for polar contribution) plus surface area model (for nonpolar contribution). To consider entropy penalty upon peptide binding, the normal mode analysis (NMA) was employed to estimate the vibrational component of the entropy. For each EGFR–MIGs2 complex system, the NMA was performed for twice; one on the MIGs2 peptide in bound state and another on unbound state. Here, the entropy effect associated with EGFR kinase domain was ignored due to its high rigidity.
Peptide-binding assay
Peptides were synthesized using standard 9-fluorenyl methoxycarbonyl (Fmoc) solid-phase chemistry. Phosphotyrosine was directly incorporated as its Nα-fluorenylmethoxycarbonyl-O-phosphate-l-tyrosine derivative. For cyclic peptide, the PAM-Gly-Boc resin was used with an S-tritylmercaptopropionic acid linker to facilitate the cyclization, as described previously (Chan et al. Citation2011). The peptides were then purified by RP-HPLC and confirmed by mass spectrometry and amino acid analysis. The fluorescence polarization (FP) assays were performed at 298 K following a protocol modified from previous reports (Park et al. Citation2013; Zhang et al. Citation2007). Synthetic peptides were labeled with N-terminally conjugated fluorescein rhodamine, which were incubated with purified protein of human EGFR kinase domain in the binding affinity assay buffer (10 mM Tris-HCl, pH 7.5, 25 mM NaCl, and 2 mM DTT) in a 96-well assay plate. The plate was mixed on a shaker for 5 min and incubated at room temperature for an additional 30 min. Polarization, defined as millipolarization units, was measured at room temperature with a fluorescence microplate reader. A negative control (5 μM peptide and assay buffer) and a positive control (5 μM peptide, 60 μM kinase domain and assay buffer) were used to determine the range of the assay. The binding was monitored as titration curves and used to derive the equilibrium dissociation constants Kd by nonlinear curve fitting: F = F0 + F∝ (p/Kd)/1 + (p/Kd), where p is the protein concentration at each measurement point, F is the observed FP value at the given protein concentration, F0 is the FP value of free peptide, and F∞ is the maximal FP value saturated with protein. Each assay was performed in duplicate.
Results and discussion
The MIG6s2 peptide is composed of 22 amino acids (376VPCILPIIENGKVCSTHYYLLP398), which is folded into two β-strands in cocrystallized complex with EGFR kinase domain (). A recent study found that phosphorylation of intact MIG6 protein at Tyr394 and Tyr395 residues is essential for binding and inhibition of EGFR (Park et al. Citation2015). The two residues are just within the MIG6s2 region, and phosphorylation renders them negatively charged that promotes the two residues to separately form two salt bridges with the positively charged residues Lys875 and Lys879 of EGFR kinase domain. According to the double-headed mechanism for EGFR inhibition by MIG6 protein proposed by Zhang et al. (Citation2007), the MIG6s1 first binds tightly to EGFR C-lobe to anchor MIG6 on EGFR, and then, the MIG6s2 is dynamic balance between association and dissociation with EGFR activation loop by phosphorylation and dephosphorylation at its Tyr394 and Tyr395 residues. Thus, it is suggested that phosphorylation is the necessary but not sufficient condition for an isolated MIG6s2 peptide binding to EGFR without MIG6s1 assistance.
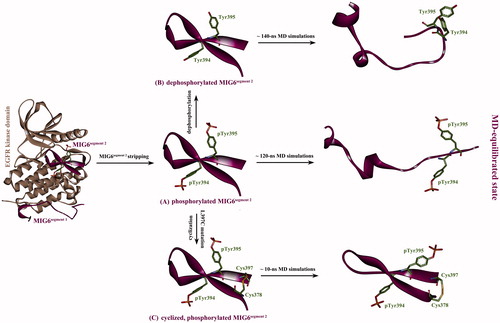
The phosphorylated MIG6s2 was striped from its cocrystallized complex with EGFR kinase domain (), and then the phosphate moieties were manually removed from the phosphorylated MIG6s2, resulting in a dephosphorylated counterpart of MIG6s2 peptide (). Subsequently, the two MIG6s2 peptides (phosphorylated and dephosphorylated) were separately subjected to a long-term MD simulation to reach at equilibrium after ∼120 ns and ∼140 ns simulations, respectively. In addition, the complex structures of EGFR kinase domain with the two peptides were also equilibrated by 50-ns MD simulations. The equilibrium state of phosphorylated and dephosphorylated MIG6s2 peptides in solution was shown in , respectively. As can be seen, there is a considerable difference between the bound and unbound conformations of the two peptides; the bound conformation is well structured that composed of two β-strands, just like that in crystal structure, while the unbound conformation exhibits typical disorder feature with two irregular helices present in N-terminal region. In fact, the intrinsic thermal motion of unbound peptides in equilibrium state is predominant and fast conformation changes can be observed during the entire MD simulations, suggesting that both the phosphorylated and dephosphorylated MIG6s2 are intrinsically disordered that cannot form specific structure in solution. According to the indirect readout theory for protein–peptide recognition proposed by Yu et al. (Citation2014), entropy penalty would be very significant upon binding of the highly flexible MIG6s2 peptide to EGFR protein. This is because peptide is highly flexible molecule that would incur considerable loss in degrees of freedom (i.e. entropy decrease ΔS < 0) upon binding to protein receptor. According to the Gibbs’ equation ΔG = ΔH – TΔS, the ΔS < 0 means –TΔS > 0, which causes ΔG increase that is unfavorable to peptide binding (ΔG < 0 and >0 represent that the binding can be spontaneous and cannot be spontaneous, respectively). A large entropy penalty associated with peptide binding to protein has been observed in crystal structure analysis of protein–peptide complexes (London et al. Citation2010) and MD simulations of Tsg101 protein interaction with an HIV-derived PTAP peptide (Killian et al. Citation2009).
Figure 2. (A) The phosphorylated MIG6s2 peptide was stripped from its cocrystallizated complex structure with EGFR kinase domain (PDB: 4ZJV), resulting in unbound phosphorylated peptide with the conformation as it in cocrystallizated complex. (B) The stripped peptide was dephosphorylated at residues pTyr394 and pTyr395 without conformation change, resulting in a dephosphorylated version of the peptide. (C) The Leu397 residue of the stripped peptide was virtually mutated to Cys397, which was then covalently connected to wild-type Cys378 residue by computationally modeling a disulfide bond between them, resulting in a phosphorylated cyclic peptide. Subsequently, the three artificially modeled peptides were subjected to MD simulations to reach at equilibrium state.
In order to clarify the effect of Tyr394 and Tyr395 phosphorylation on the intermolecular interaction between EGFR and MIG6s2 peptides, the binding free energies ΔG of both phosphorylated and dephosphorylated MIG6s2 peptides to EGFR kinase domain were calculated using MM/PBSA + NMA analysis. Based on the analysis, we can computationally derive the total binding free energy ΔG and decomposed energetic components of the two linear peptides to EGFR. The calculated energetics parameters are tabulated in . It is unraveled that: (i) phosphorylation can considerably improve the intermolecular interaction potency between EGFR kinase domain and MIG6s2 peptide (ΔEint changes from −176.4 to −240.7 kcal/mol upon the phosphorylation); this is expected because the introduced phosphates in MIG6s2 can form two electrostatically attractive salt bridges with EGFR as revealed by crystal structure. (ii) The desolvation effect is unfavorable (ΔDdslv > 0) since polar water molecules are stripped from the hydrophilic complex interface. The phosphorylation can substantially amplify the unfavorable desolvation effect (ΔDdslv increases from 115.8 to 160.9 kcal/mol upon the phosphorylation) due to the presence of charged phosphates at the interface. (iii) As might be expected, entropy penalty is as much as ∼70 kcal/mol upon the binding of highly flexible MIG6s2 peptide to EGFR, which is not different significantly between the phosphorylated and dephosphorylated peptides (–TΔS = 76.4 and 67.8 kcal/mol, respectively). Consequently, the total binding free energies ΔG of phosphorylated and dephosphorylated peptides are –3.4 and 7.2 kcal/mol, indicating that the former and the latter are weak binder and nonbinder of EGFR, respectively.
Table 1. The calculated energetics parameters and experimental affinities for the different forms of MIG6s2 peptides to interact with EGFR kinase domain.
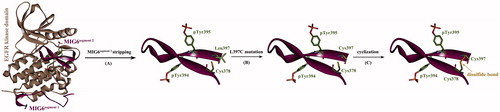
In order to improve the binding capability of phosphorylated MIG6s2 peptide to EGFR kinase domain, we herein attempted to further optimize the peptide structure to obtain increased affinity. According to above calculations, the entropy penalty is an important energetic factor unfavorable to peptide binding, which cannot be influenced significantly by phosphorylation. To minimize the entropy penalty upon peptide binding, we tried to constrain the conformational flexibility of MIG6s2 peptide in unbound state. Considering that the peptide is folded into two antiparallel β-stands in cocrystallizated complex where its C- and N-terminuses are spatially close to each other, it is assumed that the two ends of the peptide can be covalently connected by a disulfide bond to generate a cyclic peptide. A computational procedure for the cyclization of linear phosphorylated MIG6s2 peptide is shown in . First, the phosphorylated peptide with bound conformation was stripped from complex crystal structure (). By examining the conformation, it was found that a Cys378 residue is present at the peptide N-terminus, which can be considered as an “anchor” to create disulfide bond. Correspondingly, the C-terminal residue Leu397 can serve as a good candidate of another “anchor,” which is spatially vicinal to the N-terminal Cys378 residue. Here, the Leu397 residue was virtually mutated to Cys397 () using the WHAT IF server (Vriend Citation1990), and then, a disulfide bond was modeled between the mutated Cys397 and wild-type Cys378 to cyclize the peptide (). Subsequently, the phosphorylated cyclic peptide was subjected to MD simulations to relax the artificial system. As might be expected, the cyclic peptide reached at its dynamics equilibrium only taking ∼10 ns simulations, which was much fast as compared to those linear peptides (∼120 ns and ∼140 ns simulations for linear phosphorylated and dephosphorylated peptides, respectively). In addition, the MD-equilibrated conformation of the cyclic peptide is roughly consistent with that of MIG6s2 in cocrystallizated complex (), suggesting that the cyclic peptide is prestructured and its solution conformation is constrained to that in complex with EGFR. In this way, the entropy penalty upon the peptide binding to EGFR is expected to be largely minimized. This can be solidified by NMA analysis of entropy effect associated with the binding. As can be seen in , the cyclization of phosphorylated MIG6s2 peptide does not cause significant influence on the intermolecular interaction (ΔEint) and desolvation effect (ΔDdslv) of EGFR–peptide binding but can largely reduce entropy penalty upon the binding (−TΔS changes from 76.4 to 34.2 kcal/mol due to the cyclization). Consequently, the cyclic peptide exhibits a high affinity toward EGFR kinase domain with calculated binding free energy ΔG of −16.3 kcal/mol, which is much higher than that of linear peptide (ΔG = −3.4 and 7.2 kcal/mol for linear phosphorylated and dephosphorylated peptides, respectively). It is therefore expected that the cyclic peptide can bind more tightly to EGFR kinase domain than its uncyclized counterpart, the linear phosphorylated MIG6s2.
Figure 3. Cyclization of phosphorylated MIG6s2 peptide. (A) The phosphorylated peptide was stripped from its cocrystallizated complex with EGFR kinase domain (PDB: 4ZJV). (B) The Leu397 residue of the peptide was virtually mutated to Cys397 (L397C). (C) The peptide was cyclized by introducing a disulfide bond between the mutated Cys397 and wild-type Cys378.
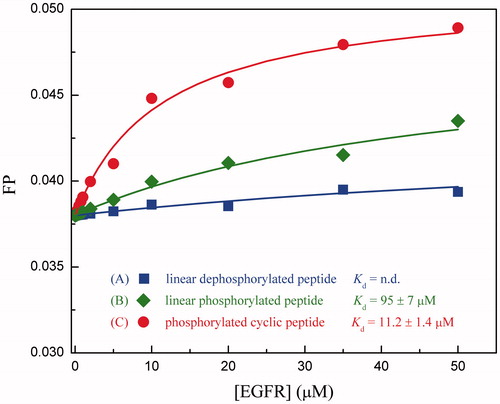
Next, we employed fluorescence polarization assay to measure the binding affinity of linear phosphorylated and dephosphorylated MIG6s2 peptides and phosphorylated cyclic peptide to the recombinant protein of human EGFR kinase domain. The titration curves are shown in . It is evident that the phosphorylated cyclic peptide impacts significantly on fluorescence emission, indicating that the peptide can bind potently to EGFR protein. The dissociation constant Kd of the peptide was determined as 11.2 ± 1.4 μM by curve fitting through the fluorescence titrations. In contract, the linear dephosphorylated peptide has no observable binding for EGFR, given a very modest fluorescence perturbation with titration of the peptide (Kd = n.d.). In addition, it was also found that the phosphorylation can effectively improve the binding potency for the linear peptide to obtain a moderate affinity (Kd = 95 ± 7 μM). In this respect, the experimental evidences are well consistent with that of computational findings, that is, the MIG6s2 peptide can be changed from nonbinder (Kd = n.d.) to weak binder (Kd = 95 ± 7 μM) and then to moderate binder (Kd = 11.2 ± 1.4 μM) of EGFR by phosphorylation and cyclization, respectively. We therefore expected that strong binder (Kd at nanomolar level) could be obtained by a further sequence optimization and structure modification of the currently designed phosphorylated cyclic peptide.
Figure 4. Titration of recombinant EGFR kinase domain to different chemical forms of fluorescein-labeled MIG6s2 peptides.
Disclosure statement
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
References
- Ai X, Sun Y, Wang H, Lu S. 2014. A systematic profile of clinical inhibitors responsive to EGFR somatic amino acid mutations in lung cancer: implication for the molecular mechanism of drug resistance and sensitivity. Amino Acids. 46:1635–1648.
- Bethune G, Bethune D, Ridgway N, Xu Z. 2010. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2:48–51.
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, et al. 2005. The amber biomolecular simulation programs. J Comput Chem. 26:1668–1688.
- Chan LY, Gunasekera S, Henriques ST, Worth NF, Le SJ, Clark RJ, et al. 2011. Engineering pro-angiogenic peptides using stable disulfide-rich cyclic scaffolds. Blood. 118:6709–6717.
- Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. Chem Phys. 98:10089–10092.
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, et al. 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 24:1999–2012.
- Ferby I, Reschke M, Kudlacek O, Knyazev P, Pantè G, Amann K, et al. 2006. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 12:568–573.
- Hackel PO, Gishizky M, Ullrich A. 2001. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 382:1649–1662.
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 79:926–935.
- Killian BJ, Kravitz JY, Somani S, Dasgupta P, Pang YP, Gilson MK. 2009. Configurational entropy in protein-peptide binding: computational study of Tsg101 ubiquitin E2 variant domain with an HIV-derived PTAP nonapeptide. J Mol Biol. 389:315–335.
- Kim TH, Yoo JY, Kim HI, Gilbert J, Ku BJ, Li J, et al. 2014. Mig-6 suppresses endometrial cancer associated with Pten deficiency and ERK activation. Cancer Res. 74:7371–7382.
- Izumchenko E, Sidransky D. 2015. Understanding the MIG6-EGFR signaling axis in lung tumorigenesis. Cancer Discov. 5:472–474.
- Li ZX, Qu LY, Wen H, Zhong HS, Xu K, Qiu XS, Wang EH. 2014. Mig-6 overcomes gefitinib resistance by inhibiting EGFR/ERK pathway in non-small cell lung cancer cell lines. Int J Clin Exp Pathol. 7:7304–7311.
- London N, Movshovitz-Attias D, Schueler-Furman O. 2010. The structural basis of peptide-protein binding strategies. Structure. 18:188–199.
- Maity TK, Venugopalan A, Linnoila I, Cultraro CM, Giannakou A, Nemati R, et al. 2015. Loss of MIG6 accelerates initiation and progression of mutant epidermal growth factor receptor-driven lung adenocarcinoma. Cancer Discov. 5:534–549.
- Moonrin N, Songtawee N, Rattanabunyong S, Chunsrivirot S, Mokmak W, Tongsima S, Choowongkomon K. 2015. Understanding the molecular basis of EGFR kinase domain/MIG-6 peptide recognition complex using computational analyses. BMC Bioinformatics. 16:103.
- Park E, Kim N, Ficarro SB, Zhang Y, Lee BI, Cho A, et al. 2015. Structure and mechanism of activity-based inhibition of the EGF receptor by Mig6. Nat Struct Mol Biol. 22:703–711.
- Park D, Magis AT, Li R, Owonikoko TK, Sica GL, Sun SY, et al. 2013. Novel small-molecule inhibitors of Bcl-XL to treat lung cancer. Cancer Res. 73:5485–5496.
- Ryckaert JP, Ciccotti G, Berendsen HJC. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 23:327–341.
- Steinbrecher T, Latzer J, Case DA. 2012. Revised AMBER parameters for bioorganic phosphates. J Chem Theory Comput. 8:4405–4412.
- Vriend G. 1990. WHAT IF: A molecular modeling and drug design program. J Mol Graph. 8:52–56.
- Wendt MK, Williams WK, Pascuzzi PE, Balanis NG, Schiemann BJ, Carlin CR, Schiemann WP. 2015. The antitumorigenic function of EGFR in metastatic breast cancer is regulated by expression of Mig6. Neoplasia. 17:124–133.
- Yu H, Zhou P, Deng M, Shang Z. 2014. Indirect readout in protein-peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model. 54:2022–2032.
- Yu XD, Yang R, Leng CJ. 2016. Truncation, modification, and optimization of MIG6(segment 2) peptide to target lung cancer-related EGFR. Comput Biol Chem. 61:251–257.
- Yang C, Zhang S, He P, Wang C, Huang J, Zhou P. 2015. Self-binding peptides: folding or binding? J Chem Inf Model. 55:329–342.
- Yang C, Zhang S, Bai Z, Hou S, Wu D, Huang J, Zhou P. 2016. A two-step binding mechanism for the self-binding peptide recognition of target domains. Mol Biosyst. 12:1201–1213.
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. 2006. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 125:1137–1149.
- Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. 2007. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 450:741–744.
- Zhang YW, Staal B, Su Y, Swiatek P, Zhao P, Cao B, et al. 2007. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene. 26:269–276.