
Abstract
The objective of this study was to compare two lansoprazole products (the reference brand enteric-coated capsules and the test generic enteric-coated tablets), and the key pharmacokinetic (PK) parameters for both formulations and their metabolites were assessed. The study used an open-label, randomized two-period crossover design with an 8-day washout period between doses in 24 healthy subjects under fasting conditions. The concentration of lansoprazole and its two metabolites was determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. The in vitro release of lansoprazole from the test and reference formulations was within the acceptable limits. The relationship between concentration and peak area ratio was found to be linear within the range for lansoprazole and metabolites. The point estimates (ratios of geometric mean) of lansoprazole and two main metabolites were between 94.3% and 105.1% for AUC0−t, AUC0−∞, and Cmax. No statistically significant difference between the two formulations was found. The results of in vitro and in vivo suggested equivalent clinical efficacy of the two brands.
Introduction
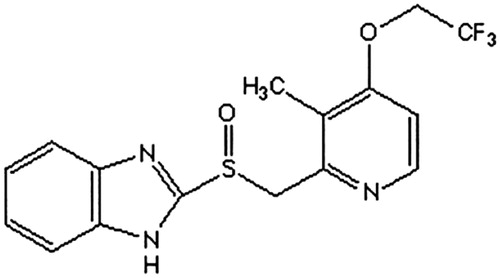
Lansoprazole is 2 {(3-methyl-4-(2,2,2-oroethoxy)-2-pyridyl) methyl} sulfinyl benzimidazole, as shown in (Dugger et al. Citation2001; Inatomi et al. Citation2008). It is a kind of benzimidazole derivatives and inhibits gastric acid secretion and antiulcer activity. Lansoprazole is transformed into active sulfonamide metabolites in the acidic environment of the gastric parietal cells; these metabolic inhibition (H+/K+)-ATPase (Gremse Citation2001; Schulz and Steinijans Citation1992). A lot of research has confirmed that oral lansoprazole, 30 mg/day, provides effective relief of symptoms and healing of duodenal ulcer in 75–100% of patients after 4 weeks of therapy in non-comparative and comparative trials (Miner et al. Citation2003; Uno et al. Citation2005; Zeng et al. Citation2015). So far, it has been widely used for eradication of Helicobacter pylori (HP) combined clinically with clarithromycin and amoxicillin (Uno et al. Citation2005).
Figure 1. The structure of lansoprazole.
Since lansoprazole is acid-labile, it is usually administered as capsules containing enteric-coated granules to prevent gastric decomposition and to increase the bioavailability (Takepron®). Lansoprazole has a half-life of about 1.5 h, an absolute bioavailability of about 80–91%, which may be decreased if administered within 30 min of food intake. The peak concentration after oral administration was within 1 ± 2 h; a single 30 mg oral dose gives a peak concentration of 750–1150 ng/mL (Alai and Lin Citation2015; Frazzoni et al. Citation2002; USP DI Citation1998; Welage and Berardi Citation2000).
In China, several commercial preparations are available now (due to the expiration of the pharmaceutical patent), and it is of critical importance to demonstrate that these preparations are bioequivalent and have similar inhibition of HP reactivity. During the development of new generic formulations, the formulators try to adhere to the same excipients used in the brand product and the manufacturing process as much as they can minimize the problems with stability. According to the International Conference on Harmonization (ICH) guidelines, the accelerated stability study was conducted. After storage stability at 40 °C and 75% relative humidity (RH), there was no any significant difference in both appearance characteristics and drug content. However, it is important to conduct an in vivo study to find any difference between the generic product and the original brand in terms of safety and clinical outcome. In additional, lansoprazole sulphone (LS) and 5′-hydroxy lansoprazole (HL) were the two main metabolites of lansoprazole. The content of metabolites also directly reflects the quality of the product.
The objective of this study was to compare two lansoprazole products (the reference brand enteric-coated capsules and the test generic enteric-coated tablets), and the key pharmacokinetic (PK) parameters for both formulations and their metabolites were assessed (Davit et al. Citation2013; Rediguieri et al. Citation2014). Thus, we determined the concentration of lansoprazole and its metabolites using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to explain the characteristics of drug metabolism in Chinese subjects. This generic is expected to be marketed in China, so it is important to test its bioequivalence in this group of people because lansoprazole is highly affected by genetic polymorphisms (Zhang et al. Citation2014).
Materials and methods
Drugs and materials
Lansoprazole 30-mg enteric-coated capsules (brand name: Takepron, batch: 150212; Taketa, Japan) and lansoprazole 30-mg enteric-coated tablets (batch: S0291; Local company, China) were used in this study. Both drugs were available by the manufacturers for free. Bicalutamide (internal standard), LS, and HL standard were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). All reagents were of high-performance liquid chromatography (HPLC) (Sigma-Aldrich, Shanghai, China) grade. Other reagents were of analytical grade. Purified water from a Milli-Q system (Millipore, Bedford, MA) was used throughout the experiment.
Volunteers
The study included 24 male healthy volunteers, aged between 18 years and 24 years, with a body mass index of 19.2–29.6 kg/m2 (minimum of 50 kg weight), as well as nonsmokers. The volunteers were subjected to a full medical and physical examination to confirm their health status and to verify that they were not on any medication during the study period for full adherence with the inclusion criteria. A written informed consent form, which explained the nature of the study, was given to the volunteers. The volunteers were instructed to abstain from taking drugs for 1 month before the study initiation, to abstain from caffeine and alcohol-containing beverages for at least 16 h prior to each study drug administration and throughout the study period, and fast for at least 10 h before drug administration. Each volunteer completed an acceptable medical history, medication history, physical examination, electrocardiogram examination, screening of HIV-1 and 2 antibody and hepatitis B surface antigen, and urine drug screening studies before the start of the study. Selection of routine clinical laboratory measurements for screening. Upon completion of the study, physical examination and clinical laboratory measurements were repeated.
The study was conducted in accordance with the requirements of the Declaration of Helsinki (World Medical Association Citation2008) and the International Conference on Harmonization guidelines (Barrett et al. Citation2000; ICH Citation1996).
Study design
The study used an open-label, randomized two-period crossover design with an 8-day washout period between doses in 24 healthy subjects under fasting conditions. The volunteers were randomly divided into two groups each of 12 subjects. The first group was given the reference formulation, and the second group was given the test formulation with a crossover after the washout period.
On the morning of the study, a catheter was placed in a suitable forearm vein of each volunteer and a 5-mL blood sample serves as a blank for the drug assay. Each volunteer access to water was not restricted until 1 h before administration (during the fasting period). And then they received an oral dose of the assigned formulation given with 240 mL of water in the sitting position. Additional blood samples (5 mL) were obtained by individual venupuncture at designated time points (pre-dose and 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h after administration) and were transferred into tubes. The plasma was separated by centrifugation at 4000 × g (10 min) and stored at −20 °C until further analysis. Four hours after drug administration, a standard lunch meal was served, and volunteers had free access to water 1 h after drug administration.
Instruments and chromatographic separations
LC-MS/MS method was modified according to the previous reports (Oliveira et al. Citation2003; Wu et al. Citation2008; Zhang et al. Citation2013). Chromatography was performed using an API4000 tandem mass spectrometer (Applied Biosystems) equipped with a binary and a quaternary pump (Agilent series 1200) and with separation on a 5-μm ODS-3 column (100 mm ×4.6 mm). The mobile phase consisted of methanol and water (70:30, v/v, containing 5 mM ammonia formate, pH was adjusted to 7.5 by 1% ammonia solution) at a flow of 0.4 mL/min, and the column temperature was maintained at 25 °C.
The compounds were ionized in the negative ion polarity mode. The source/gas-dependent parameters were as follows: curtain gas, 10 psi; collision gas, 4 psi; medium temperature, 350 °C; ion spray voltage, 4000 V; ion source gas one and gas two, 25 psi; drying gas flow, 10 L/min; nebulizer pressure, 45 p.s.i; and quadrupole temperature, 105 °C. For collision-induced dissociation (CID), high purity nitrogen was used as collision gas at a pressure of 0.1 MPa. Quantification was performed using multiple reaction monitoring (MRM) mode at m/z 368.2 → 164.1 for lansoprazole, m/z 429.1 → 255.0 for bicalutamide (IS), m/z 384.0 → 115.9 for LS, and m/z 384.0 → 180.0 for HL. The fragmentation energies of Q1 for the analytes were set at 120 V. The optimized collision energies of 20 and 10 eV were used for lansoprazole and bicalutamide, respectively. During the data acquisition, the delta potential of the electron multiplier voltage (EMV) was set to 200V. The peak widths of precursor and product ions were maintained at 0.7 amu at half-height in the MRM mode.
Sample preparations
100 μL of plasma was added to 1.5-mL plastic test tube containing the internal standard (bicalutamide, 20 μL of 0.5 μg/mL), and 300 μL of acetonitrile was then added. After vigorous vortex mixing for 30 s (Eppendorf, 5432 vortex mixer, Hamburg, Germany), the mixture was centrifuged for 10 min at 4000 × g. The organic phase was transferred to a clean vial, and a 5 μL aliquot of the solution was injected into the LC-MS/MS system for analysis.
Validation procedures
The precision and accuracy of the assay were obtained by comparing the predicted concentration (obtained from the calibration curve) to the actual concentration of lansoprazole, LS, and HL spiked in blank plasma. Intra-day precision was measured by repeated analysis of each quality control (QC) sample on 1 day (n = 5), and inter-day precision was measured by repeated analysis for five consecutive days (1 series per day). The precision was expressed as the inter-day and intra-day coefficient of variation [CV = (SD/average recovery rate) × 100%]. Accuracy was defined as the relative deviation in the computed value (E) of a standard from that of its true value (T) expressed as a percentage (RE%). It was calculated using the formula RE% = (E − T)/T × 100. The lower limit of detection (LLOD) for lansoprazole, LS, and HL was considered as the concentration that produced a signal-to-noise (S/N) ratio of 10.
The standard solution was added to the blank plasma of three QC levels, and the freeze and thaw stability study samples were obtained. These samples were frozen at −20 °C for 30 days, and then thawed at room temperature. After being allowed to completely thaw, the samples were refrozen for 24 h under the same conditions. Before these samples were analyzed, this freeze–thaw cycle was repeated three times.
The absolute recovery of lansoprazole, LS, and HL was determined by direct comparison of peak areas from extracts versus spiked post-extraction samples at three QC levels, respectively. (lansoprazole (50, 750, 1500 ng/mL), LS (10, 50, 125 ng/mL), and HL (5, 25, 50 ng/mL)).
In vitro dissolution profile
In vitro dissolution study was carried out on six tablets or capsules from test and reference formulations. After immersed in acidic medium (0.1MHCl) for 1 h, the drug release from the formulation was measured in a paddle USP apparatus (75 rpm, 37 °C, 900 mL PBS (pH 6.8)). At specific time intervals, samples were withdrawn and analyzed using an HPLC assay described below.
Safety evaluations
Safety profile evaluations were conducted through monitoring of vital signs that included sitting blood pressure, pulse, respiratory rate, oral temperature, and body weight. The subjects were closely monitored for evidence of drug intolerance and for the development of clinical or laboratory evidence of adverse events. All adverse events that occurred during the study were reported in detail on the subjects’ charts, and follow a satisfactory resolution. The researchers assessed the severity of each adverse event and the possible relationship with the study drug. Severity was defined as follows: mild, transient, and easily tolerated; interrupt moderate, causes the body discomfort and normal activities; or serious, resulting in normal activities of considerable interference, and disabling or life-threatening. In addition, relationship to study drug was determined as definite, probable, possible, or not related.
Pharmacokinetic
Data for the concentration in plasma versus time for both test and reference formulations were analyzed with noncompartmental methods (WinNonlin® (Version 5.0.1). PK variables of interest included area under the concentration-time curve from time 0 to t (AUC0−t) and time 0 to ∞ (AUC0−∞), peak concentration (Cmax), terminal elimination rate constant (Ke), half-life of the terminal phase (t1/2), and time to peak concentration (Tmax). A linear-up/log down method of estimation was used for the calculation of AUC0−∞ and was obtained by adding Clast/Ke to AUC0−t. The terminal elimination rate constant (Ke) was determined from the slope of terminal exponential phase of the logarithmic plasma concentration-time curve. The elimination half-life (t1/2) was calculated using 0.693/Ke.
Statistical analysis
For the purpose of bioequivalence analysis AUC0−t, AUC0−∞, and Cmax were considered as primary variables. The biological equivalence is the 90% confidence interval of the ratio test/reference (T/R) by means of an analysis of variance (ANOVA model evaluation method) for crossover design and calculation standard. The products were considered bioequivalent if the difference between two compared parameters was found statistically significant (P < .05) and 90% confidence intervals for these parameters fell within 80 ± 120%. The acceptance range of Cmax may be wider than AUC, especially with highly variable peak concentrations; the recommended range is 70–143%.
Results
Method validation
For the construction of calibration curves with spiked samples, the method of constant addition was used. The calibration curves of the corrected peak areas versus concentration (ng/mL) were linear in the range studied. The data from the calibration curve are summarized in . LLOD values were determined as the concentration giving a peak height 10 times the noise background. The intra-/inter-assay precision and accuracy indicated that RSDs of intra-assay were between 6.87% and 10.52% for lansoprazole, 7.17% and 12.13% for LS, and 6.72% and 9.89% for HL, respectively. The inter-assay RSDs were between 8.78% and 13.27% for lansoprazole, 7.26% and 12.43% for LS, and 7.82% and 11.52% for HL, respectively. The overall mean recoveries for lansoprazole, LS, and HL at low quality control (LQC), middle quality control (MQC), and high quality control (HQC) levels were nearly 100% and with RSD% between them of 3.63% for lansoprazole, 4.72% for LS, and 7.21% for HL. Thus, the consistency in recoveries supported the extraction procedure for its application to routine sample analysis.
Table 1. Linearity data (line equation with a standard calibration curve) in spiked samples of blank plasma (n = 6 for each concentration level).
Three freeze–thaw cycles demonstrated that about 91.1%–93.2% of the initial content of lansoprazole, LS, and HL were recovered and that the analytes were stable under these conditions. Plasma samples collected from studies of lansoprazole, LS, and HL were evaluated before and after storage at −20 °C for stability and found to be stable for at least 30 days.
In vitro dissolution profile
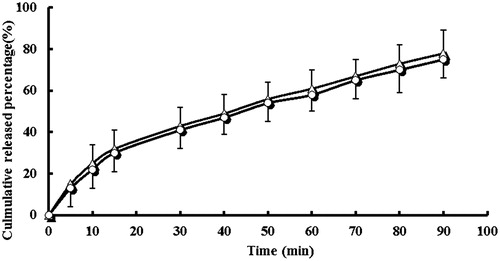
In vitro drug-release studies were carried out because these are considered a useful tool for assessing in vivo drug behavior. The in vitro release of lansoprazole from the test and reference formulations was within the acceptable limits. The calculated f2 values between these two products were 58 ().
Figure 2. In vitro dissolution profile of lansoprazole tablets (△) and lansoprazole capsules (^) in the medium of 900 mL PBS (pH 6.8) (n = 6).
Pharmacokinetic
Mean plasma concentration curve of lansoprazole, LS, and HL by following administration of lansoprazole tablets and lansoprazole capsules were shown in . The plasma concentrations of both brands indicated that the two brands are superimposable. The PK parameters and statistical evaluation of lansoprazole, LS, and HL were summarized in . All estimated PK parameters were in agreement with reported values (Oliveira et al. Citation2003; Wu et al. Citation2008; Zhang et al. Citation2013). showed a summary of the PK parameters for the two formulations of lansoprazole, LS, and HL. The point estimates (ratios of geometric mean) of lansoprazole and two main metabolites were between 94.3% and 105.1% for AUC0−t, AUC0−∞, and Cmax. No statistically significant difference between the two formulations was found. These PK parameter values lie within the bioequivalence limits (80–125%) specified by the Food and Drug Administration (FDA) and the European Medicines Agency (EMEA). The results in this part of the study suggest equivalent clinical efficacy of the two brands.
Figure 3. Mean plasma concentration curve of lansoprazole by following administration of tablets (^) and capsules (△) (n = 12, single dose: 30 mg).
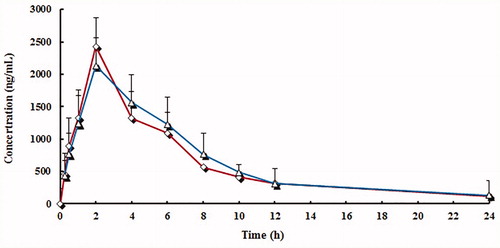
Figure 4. Mean plasma concentration curve of lansoprazole sulphone (LS) by following administration of tablets (^) and capsules (△) (n = 12, single dose: 30 mg).
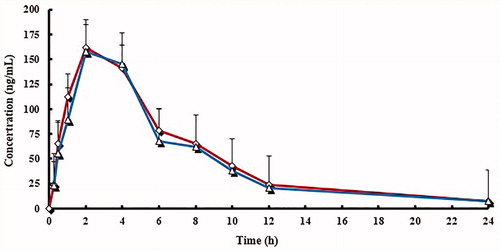
Figure 5. Mean plasma concentration curve of 5′-hydroxy lansoprazole (HL) by following administration of tablets (^) and capsules (△) (n = 12, single dose: 30 mg).
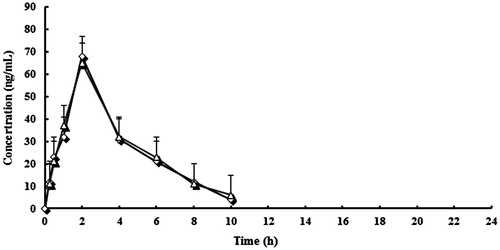
Table 2. Pharmacokinetic parameters of lansoprazole of the two formulations (n = 12, mean ± SD).
Table 3. Pharmacokinetic parameters of lansoprazole sulphone (LS) of the two formulations (n = 12, mean ± SD).
Table 4. Pharmacokinetic parameters of 5′-hydroxy lansoprazole (HL) of the two formulations (n = 12, mean ± SD).
Discussion
According to FDA, a brand name drug is defined as a drug marketed under a proprietary, trademark-protected name and a generic drug is the same as a brand name drug with regard to active ingredient, dosage form, safety, strength, route of administration, quality, performance as assessed from the PK profile, and intended use and contains the same salt, ester, or chemical form. Generic versions of a drug can vary in shape, scoring configuration, packaging, and excipients. If all the previous criteria are met, then the two drugs are considered to be equivalent to the treatment (Guideline … in the paediatric population Citation2009).
During the development phase of generic formulation, several preformulation and formulation trials and tests are carried out to achieve a generic product that can be interchangeable with the original brand in terms of efficacy and safety. Accordingly, in vitro dissolution is conducted on the generic product, and it must show a similar dissolution profile or overlap with the reference brand. In this study, the in vitro release of lansoprazole from test and reference formulations was within the acceptable limits. Of course, in vitro release cannot replace the in vivo tests that demonstrate the efficacy and safety of the generic product since it may contain different excipients that affect the rate of release in vivo.
Regarding the efficacy of our generic product, statistical comparison of the main PK parameters, AUC0−t, AUC0−∞, and Cmax, clearly indicated no significant difference between test and reference formulations. The values obtained were compliant with the FDA and EMEA requirements for bioequivalence of generic drugs since the AUC0−t, AUC0−∞, and Cmax mean ratios are within the 80–125% interval.
Safety is also important. Administration of a single oral dose of either the test or reference formulation was found to be safe in healthy subjects. No severe, serious, or life-threatening clinically or drug-related side effects were reported throughout the study. Accordingly, this generic product offers an efficacious treatment option for individual patient with relatively lower cost in comparison with the established brand drug.
Disclosure statement
All authors have no conflicts of interest to disclose.
References
- Alai M, Lin WJ. 2015. Application of nanoparticles for oral delivery of acid-labile lansoprazole in the treatment of gastric ulcer: in vitro and in vivo evaluations. Int J Nanomedicine. 10:4029–4041.
- Barrett JS, Batra V, Chow A, Cook J, Gould AL, Heller AH, et al. 2000. PhRMA perspective on population and individual bioequivalence. J Clin Pharmacol. 40:561–570.
- Davit B, Braddy AC, Conner DP, Yu LX. 2013. International guidelines for bioequivalence of systemically available orally administered generic drug products: a survey of similarities and differences. AAPS J. 15:974–990.
- Dugger HA, Carlson JD, Henderson W, Erdmann GR, Alam SM, Dham R, et al. 2001. Bioequivalence evaluation of lansoprazole 30-mg capsules (Lanfast and Lanzor) in healthy volunteers. Eur J Pharm Biopharm. 51:153–157.
- Frazzoni M, De Micheli E, Grisendi A, Savarino V. 2002. Lansoprazole vs. omeprazole for gastro-oesophageal reflux disease: a pH-metric comparison. Aliment Pharmacol Ther. 16:35–39.
- Gremse DA. 2001. Lansoprazole: pharmacokinetics, pharmacodynamics and clinical uses. Expert Opin Pharmacother. 2:1663–1670.
- Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population, 2009. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf.
- ICH. 1996. ICH harmonization tripartite guideline. Guidelines for good clinical practice. International Conference of Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human uses, Geneva, Switzerland.
- Inatomi N, Nitta K, Sakurai Y. 2008. Pharmacological and clinical profile of lansoprazole (Takepron IV injection 30 mg). Nihon Yakurigaku Zasshi. 131:149–156.
- Miner P, Jr, Katz PO, Chen Y, Sostek M. 2003. Gastric acid control with esomeprazole, lansoprazole, omeprazole, pantoprazole, and rabeprazole: a five-way crossover study. Am J Gastroenterol. 98:2616–2620.
- Oliveira CH, Barrientos-Astigarraga RE, Abib E, Mendes GD, da Silva DR, de Nucci G. 2003. Lansoprazole quantification in human plasma by liquid chromatography-electrospray tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 783:453–459.
- Rediguieri CF, Cristofoletti R, Soares KC, Tavares-Neto J. 2014. Similarities and differences of international guidelines for bioequivalence: an update of the Brazilian requirements. AAPS J. 16:350–351.
- Schulz HU, Steinijans VW. 1992. Striving for standards in bioequivalence assessment: a review. Int J Clin Pharmacol Ther Toxicol. 30:S1–S6.
- Uno T, Yasui-Furukori N, Takahata T, Suqawara K, Tateishi T. 2005. Determination of lansoprazole and two of its metabolites by liquid-liquid extraction and automated column-switching high-performance liquid chromatography: application to measuring CYP2C19 activity. J Chromatogr B Analyt Technol Biomed Life Sci. 816:309–314.
- USP DI, 18th Edition, Lansoprazole, Vol. 1, United states Pharmacopoeial Convention Inc., Rockville, MD, 1998, pp. 1795–1797.
- Welage LS, Berardi RR. 2000. Evaluation of omeprazole, lansoprazole, pantoprazole, and rabeprazole in the treatment of acid-related diseases. J Am Pharm Assoc. 40:52–62.
- World Medical Association. 2008. “Declaration of Helsinki” as Amended by the 59th World Medical Assembly. Seoul, South Korea: World Medical Association Inc.
- Wu GL, Zhou HL, Shentu JZ, He QJ, Yang B. 2008. Determination of lansoprazole in human plasma by rapid resolution liquid chromatography-electrospray tandem mass spectrometry: application to a bioequivalence study on Chinese volunteers. J Pharm Biomed Anal. 48:1485–1489.
- Zeng Y, Ye Y, Liang D, Guo C, Li L. 2015. Meta-analysis of the efficacy of lansoprazole and omeprazole for the treatment of H. pylori-associated duodenal ulcer. Int J Physiol Pathophysiol Pharmacol. 7:158–164.
- Zhang D, Zhang Y, Liu M, Wang X, Yang M, Han J, et al. 2013. Pharmacokinetics of lansoprazole and its main metabolites after single and multiple intravenous doses in healthy Chinese subjects. Eur J Drug Metab Pharmacokinet. 38:209–215.
- Zhang YX, Wei SJ, Yang XY, Zhang WP, Wang XY, Dang HW. 2014. Effects of genetic polymorphisms of CYP2C19*2/*3 and MDR1 C3435T on the pharmacokinetics of lansoprazole in healthy Chinese subjects. Int J Clin Pharmacol Ther. 52:850–855.