
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The present study was designed to investigate the tumor-targeting potential of gemcitabine (GEM)-loaded surface-tailored chitosan (CS)/poly (ethylene glycol) nanoparticles (FA-PEG-GEM-NPs). The nanoparticles encapsulated with GEM were prepared, characterized, and tethered with folic acid. The developed formulations were characterized with respect to particle size/poly-dispersity index, shape, and zeta potential analysis. The in vitro study shows the sustained drug-release kinetics during 48 h. The present result shows remarkable cytotoxicity rendered by GEM when delivered through FA-PEG-GEM-NPs formulation. The microscopic assessment is suggestive of significant uptake of FA-PEG-GEM-NPs in comparison with the unmodified PEG-GEM-NPs and free drug. Finally, our results advocate for the sizeable compatibility, comparatively less organ toxicity, and higher anti-tumor activity of ligand-anchored and PEGylated CS nanoparticles in vitro and corroborated by in vivo investigations. In conclusion, it is interpreted that surface-tailored nanoparticles are capable to ferry bioactives selectively and specifically to tumor sites with the interception of minimal side effects, thereby suggesting their potential application in cancer therapeutics.
Introduction
Lung cancer is one of the most prominent forms of cancer and plays a crucial role with greater rate of mortality in both primary and metastases neoplasia (Jemal et al. Citation2004, Kerr Citation2001). In all the lung cancers, non-small-cell-lung-cancer (NSCLC) is more important. The patients diagnosed with NSCLC do not have any chemoprophylaxis. The conventional chemotherapy is associated with adverse reactions that render greater toxicity to normal cells/tissues in the prognosis of lung cancer (Mitra et al. Citation2001). The platinum-based combination chemotherapy is among the first-line treatments to address the advanced NSCLC (Fruh et al. Citation2008). The toxicities such as nausea/vomiting, nephrotoxicity, and neurotoxicity refrain patients to take platinum-based chemotherapy. Therefore, third-generation chemotherapeutic agents such as Taxanes, gemcitabine (GEM), and vinorelbine may prove to be promising treatment to confer protection against NSCLC (Hsu et al. Citation2008). GEM (20,20-difluorodeoxycytidine), a nucleoside-based analogue, has a significant antitumor activity against a variety of cancers such as cancers of the pancreas, lung, ovary, and breast. The rapid metabolism and shorter half-life of drug required higher dose and repeated dosing schedule which consequently results in higher toxicity. Thus, it is needed to design a vector which can reduce the burden of frequent dosing and higher toxicity associated with high dosages of GEM (Nandini et al. Citation2015, Sandler et al. Citation2000). Therefore, the novel drug delivery strategies are required to increase the bioavailability of drugs and to reduce side effects.
Chitosan (CS) is a natural polymer with excellent characteristics for use in drug delivery like biodegradable, biocompatible, pH sensitive (Wang et al. Citation2013) cationic polymer with low toxicity, muco-adhesive properties, and biodegradability. It is reportedly capable to enhance the penetration of large molecules across the mucosal surfaces and specifically deliver targeted molecule in intracellular compartments (Yu et al. Citation2015). The hydrophilicity was conferred on the NPs by coating with polyethylene glycol (PEG) as an additional coating polymer to form PEG-coated (PEG-GEM-NPs) nanoparticles. Further, the chemical modification of CS NPs using PEG improves the biocompatibility of CS and reduces the adsorption (Chen et al. Citation2015, Lee et al. Citation2015) of circulating plasma proteins on the material surface (Chen et al. Citation2015).
The active targeting of drugs towards the cancer cells is widely used strategy, in which ligand-anchored drug-loaded NPs can directly interact with receptors present or overly expressed by cancer cell surfaces (Chen et al. Citation2015, Wang et al. Citation2013). There are reports that have shown that various surface-modified CS NPs have been developed for an effective, targeted, and controlled delivery of drug (Bertrand et al. Citation2014, Kesharwani and Iyer Citation2015, Koo et al. Citation2005, Moghimi et al. Citation2001). Some receptors like folate and glycosyl-phosphatidylinositol-anchored cell surface receptors are overly expressed on a majority of cancer tissues against limited expression on healthy deep-seated tissues and organs (Ji et al. Citation2015). The folate receptors (38 kDa membrane glycoprotein) are highly expressed in epithelial, ovarian, cervical, breast, lung, kidney, colorectal, and brain tumors (Zwicke et al. Citation2012). Folate receptor (FR) is present in three different isoforms: two GPI-anchored membrane proteins (FR-α and FR-β) and soluble form called FR-γ (Chen et al. Citation2014). The FR-α expression plays a pivotal role in NSCLC which was confirmed through immunohistochemistry study. The FR expression provided an in-depth understanding for the prognosis of lung adenocarcinoma patients (O'Shannessy et al. Citation2012).
The present study focuses on the development, characterization, and evaluation of anticancer activity of folic acid-conjugated and PEGylated GEM-loaded CS NPs. The overly-expressed FA receptors by the experimentally induced tumor in mice were assessed for the efficacy of this surface-engineered vehicle. The in vitro findings were corroborated by in vivo investigation to validate our results. The present study opens new vistas in the drug delivery research to develop understanding on lung cancers
Material and methods
Materials
GEM, CS LMW (∼35,000) > 90% de-acetylated and PEG were purchased from Sigma Aldrich, St. Louis, MO. Thiamine pyrophosphate (TPP) and sodium borohydride (NaBH4) were purchased from Merck (Kenilworth, NJ). Folic acid was purchased from Himedia, Mumbai, India. Mobile phase solvents were of HPLC grade, and other solvents and chemicals used were of analytical grade.
Animal ethics committee approval
All animal procedures were carried out in compliance with Department of Respiratory Medicine, Liaocheng People’s Hospital regulations. All procedures were reviewed and approved by Research Review and Ethics Board Liaocheng People’s Hospital, China.
Preparation of GEM-loaded CS nanoparticles (GEM-NPs)
Drug-loaded CS nanoparticles were developed by using the method reported earlier (Rampino et al. Citation2013) with slight modification. Briefly, LMW CS was dissolved in 0.1% (v/v) acetic acid solution (3 mg/ml), with continuous stirring for 24 h. Drug was dissolved at the same stage in the same solution, i.e. 0.1% acetic acid. pH was maintained at 5.5 by using 0.2 M sodium hydroxide solution, and further dilution was done with de-ionized water to achieve the final desired concentrations. TPP was dissolved in de-ionized water to a final concentration of 0.25 mg/ml. After this, TPP solution was incorporated into the CS solution containing 5 mg/ml GEM. The resulting NPs were allowed to stir for 3–4 h, and centrifuged at 15,000 rpm in order to separate NPs. Excess of TPP and un-encapsulated GEM was removed by washing the NPs with distilled water. The supernatants were collected together for the estimation of un-entrapped GEM.
Synthesis of FA-PEG-GEM-NPs
Synthesis of FA-conjuagated and PEGylated GEM-NPs were prepared by the method reported elsewhere (Chen et al. Citation2014). The obtained gem-NPs pellet after centrifugation was re-dispersed in Milli-Q water and 50 mg of sodium borohydride (NaBH4) was added to turn the C = N bond of CS into C − N. Thereafter, the NPs were dispersed in hydrochloric acid (HCl, 1 M) for 12 h prior to dialysis against water over-night at room temperature to eliminate non-reactive reactants. The solution was subsequently freeze-dried to obtain the fluffy powder of GEM-NPs. For further use, the GEM-NPs were re-dispersed in phosphate-buffered solution (PBS). Methoxy-PEG propionic acid (mPEG − SPA, 40 mg) was added to GEM-NPs (5 ml, 8 mg/ml) and the reaction mixture was stirred for 3–4 h at room temperature to obtain PEG − GEM-NPs. FA (1 ml, 5 mg/ml) was added to PEG − GEM-NPs (5 ml, 8 mg/ml) in the presence of catalyst, 1-ethyl-3–(3-dimethyllaminopropyl) carbodiimide hydrochloride (EDC, 25 mg) and stirring was continued for 1 h at room temperature to get yellow-colored FA-PEG-GEM-NPs. The deposits were washed with water and centrifuged three times at 15,000 rpm for 20 min each until the state in which we scarcely estimated FA in supernatant by UV spectroscopy at 303 nm using UV-2500 spectrometer.
Characterization of NPs
Particle size, poly-dispersity index (PDI), and zeta potential
Malvern zetasizer (Malvern Inc., Malvern, UK) was used for the determination of particle size, PDI, and zeta potential. The diluted suspension of NPs (10–20 times) was used for the measurement of size and PDI, while for zeta potential measurement, undiluted samples were used. The % encapsulation efficiency (EE%) of GEM-loaded CS NPs was determined by indirect method. Briefly, the supernatant was used for the determination of unentrapped GEM by using RP-HPLC method (Singh et al. Citation2015). The separations were done on C18 column (Luna C18, 150 × 4.5 mm, 5 μl) operated at 30 °C. The mobile phase consisted of ACN and water at 20:80 ratios, and the detection wavelength was set at 275 nm. The flow rate was kept constant at 1 ml/min, and the % EE was determined by the following formula:
Transmission electron microscopy (TEM) was used to study the shape and the surface morphology of NPs. Briefly, a drop of diluted aqueous suspension of NPs was placed on a membrane-coated grid surface. On to the surface of grid, a drop of 1% phosphotungstic acid was added immediately. One minute later, the excess fluid was removed, and the grid surface was air dried at room temperature and was examined under the microscope (FEI Morgagni 268 D, FEI, Dawson Creek Drive Hillsboro, OR).
In vitro studies
In vitro drug release
The drug release behavior of GEM from NPs was assessed in vitro by using dialysis bag method. PBS (10 mM, pH 5.8, and 7.4) was used as the release medium. The dialysis membrane was activated by soaking the min hot water for 12 h before use. Two milliliter of nano-suspension was dispersed in PBS and both the ends were tied-up. The dialysis bags (cut off 2000 Da) were then placed in a glass beaker containing 30 ml dissolution medium and stirred at 150 rpm. The temperature of the beaker containing the release medium was maintained at 37.0 ± 0.5 °C throughout drug-release experiments. At predetermined time intervals, 500 μl of samples were withdrawn and the release medium was then replenished with the same amount of PBS. The samples were then analyzed for drug content using RP-HPLC. Further, an equal volume of PBS (maintained at same temperature) was added after the samples were withdrawn to ensure the constant sink conditions.
Cytotoxicity assay
MTT assay was used for studying the anti-proliferative effect of FA-PEG-GEM-NPs using lung epithelial cancer cell lines (A549 cells) (Chen et al. Citation2014). A549 cells were stored in DMEM medium (Himedia, Mumbai, India) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% streptomycin, 3 mM glutamine in a 37 °C humidified incubator, and 5% CO2 atmosphere. Exponentially growing A549 cells were seeded at 3 × 104 cells/ml in 96-well plates (Sigma, Aachen, Germany). The cells were individually treated with free GEM and PEG-GEM-NPs and FA-PEG-GEM-NPs which are incubated under controlled environment (37 °C humidified incubator and 5% CO2) for 72 h. Further, the MTT solution (5 mg/ml) was added to each single well and incubated for 4 h at 37 °C to facilitate the reduction of MTT by viable cells with the formation of purple formazan crystals. The formazan crystals were dissolved in DMSO, and the absorbance of individual wells was read at 570 nm.
Cell uptake study
Fluorescent dye (FITC)-loaded CS NPs (FITC-NPs) were prepared following the same procedure as mentioned in section Preparation of GEM-loaded CS nanoparticles (GEM-NPs), but replacing the drug with FITC dye (400 μg) to the CS solution before the formation of NPs. The incorporated FITC acts as NPs and is a sensitive method to determine qualitative and quantitative cellular binding.
The cell uptake study was carried out with the slight modifications in the previously reported procedure (Jain et al. Citation2015, Tseng et al. Citation2007). Briefly, A549 cells were seeded in a T-25 flask, were grown to attain 80% confluency, and were treated with modified as well as unmodified nanoparticles at a particle concentration of 100 mg/ml. After incubation for 3 h, the cells were washed twice with PBS, trypsinized, centrifuged, and resuspended in PBS buffer. Analysis of particle uptake was done using Becton Dickinson, FACScan flow cytometer system (BD, Franklin Lakes, NJ). Simultaneously, inhibition studies were carried out and cells were pre-incubated for 2 h with folic acid (1 mg/ml), well-known high-affinity ligand for the folic acid receptor, in a complete medium. Further, NPs uptake study was carried out as described above.
In vivo study
Pharmacokinetic studies
Before starting actual animal studies, all the animal protocols were approved by the Research Review and Ethics Board Liaocheng People’s Hospital, China. Balb/c mice (4–5 weeks old, weight 25 ± 2 g) were procured from Liaocheng People’s Hospital, Liaocheng, China. The pharmacokinetics study was conducted by following the previously reported procedure (Nandini et al. Citation2015) with slight modifications. Four animals each were divided into three groups. Group I was administered with plain GEM, Group II was administered with PEG-GEM-NPs, and Group III was administered with FA-PEG-GEM-NPs with a dose equivalent to 6 mg/kg of free drug through lateral tail vein. Blood samples (200 μl) were collected at different time points through the retro-orbital plexus, and the plasma was separated by centrifuging the blood samples at 8000 g for 5 min. Plasma samples thus obtained were stored at −20 °C until use. Glacial acetic acid (50 μl) was added to plasma samples to decrease hydrogen bonding between nucleosides and proteins. Acetonitrile (0.7 ml, HPLC grade) was added to precipitate the vortex-mixed plasma proteins and centrifuged at 9000 g for 15 min at 4 °C. The supernatant was removed and collected in a glass tube and ACN (1 ml) was added to the pellet. Three cycles of vortex mixing and centrifugation procedure were carried out. The supernatants were pooled, evaporated to dryness under nitrogen flux at 42 °C (thermo-stated water bath), and stored at −80 °C. Prior to RP-HPLC analysis, residue was re-suspended in water (1 ml, HPLC grade), incubated for 5 min at 37 °C, and then centrifuged at 12,000 g for 15 min at 20 °C. Further, supernatant was removed, filtered through a 0.22 μm syringe filter, and placed in 4 ml HPLC glass vials for analytical determination. Detection of gemcitabine in serum was carried out using a RP-HPLC method as mentioned in section Preparation of GEM-loaded CS nanoparticles (GEM-NPs). Other pharmacokinetic parameters from drug serum level were determined by Kinetica software version 5 (Jain et al. Citation2015).
Bio-distribution study
Bio-distribution studies were carried out on tumor-bearing female Balb/c mice. The protocols were prepared by referring the procedures reported elsewhere (Fukuda et al. Citation2000, Jain et al. Citation2011, Kumar et al. Citation2012). Briefly, A549 cells were suspended in DMEM medium and these cells were subsequently injected intravenously into BALB/C nude (null/null) mice through lateral tail vein. Once the tumors were experimentally induced (8–10 weeks post-injection), mice were used for bio-distribution study. Animals were divided into three groups of nine mice each group for free GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs. Plain GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs were administered through lateral tail vein in a dose equivalent to 15 mg/kg body weight. Three animals from each group were sacrificed at different predetermined time points such as 2, 8, and 24 h, and various organ masses such as hearts, livers, spleens, and kidneys were collected and washed with saline, and weighed prior to homogenization in saline. Tissue samples were put on ice after homogenization procedure; the homogenate was then centrifuged at 10,000 g for 10 min. ACN was then added to supernatant to precipitate un-wanted proteins; samples were centrifuged (10,000 g, 10 min) as mentioned above. The aliquots were assayed for GEM levels using RP-HPLC to estimate the total amount of GEM.
Assessment of anti-tumor activity in vivo
Tumors were experimentally induced in the mice as discussed above. Once substantial solid mass of tumors have been developed (an average volume of ∼290 mm3), mice were divided into four groups of three mice each. Group I served as control, and Groups II, III, and IV were injected with GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs, respectively, with a dose equivalent to 15 mg/kg body weight on days 0, 2, 6, and 12 through tail vein. Mice were monitored regularly for the changes in tumor size. The size of tumor masses was measured with vernier calliper, and tumor volumes were calculated according to the following formula: V = 0.5 × a × b2, where a and b are the long and the short diameter of tumor, respectively.
Background mouse strain was administered with plain GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs, a dose equivalent to 6 mg/kg body weight through lateral tail vein (Jain et al. Citation2010). Tissue samples were kept on ice after homogenization procedure; the homogenate was then centrifuged at 10,000 g for 10 min, ACN was added to the supernatant to precipitate unwanted proteins; samples were centrifuged (10,000 g, 10 min). The aliquots were assayed for GEM levels using RP-HPLC to estimate the total amount of GEM.
Toxicity profiling
Systemic nephro-and-hepatotoxicity
The samples previously collected for pharmacokinetic study were used to assess liver and kidney functioning. The animals were injected normal saline and deemed as the control group. The urea concentration in the serum was quantified by urease glutamate dehydrogenase method and creatinine by modified Jaffe's method using the diagnostic kits of Agappe Diagnostic, India Pvt. Ltd., Kochi, Kerala (Garg et al. Citation2012). Serum activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were evaluated using commercially available test kits from Randox Laboratories Ltd. (County Antrim, UK) (Jain et al. Citation2010).
Results and discussion
Preparation and characterization of NPs
The results of the particles size suggest that the attachment of PEG and FA onto the surface of NPs resulted into an increase in the particle size of the GEM-NPs indicating the interactive association of PEF and FA on the surface. Further, it was observed that the addition of higher concentration of FA-PEG resulted in higher particles diameter and molecular weight (data not shown). It was earlier reported that the NPs with diameters larger than 200 nm have shown an induction of non-specific scavenging by monocytes and reticuloendothelial system (Na et al. Citation2003). Since average particles’ size of the formulations containing 10 mg/ml FA-PEG was found to be approx. 185 nm, FA-PEG-GEM-NPs formulation was selected for further studies to ensure reduced toxicity and better tumor target ability. We obtained a homogeneous NPs dispersion () with smooth surface (. Also, the PDI of NPs before and after lyophilization was calculated to be <0.2, which is an indication of homogenously distributed particles. Moreover, lyophilized formulation demonstrated more or less similar particle size upon reconstitution.
Figure 1. High-resolution transmission electron microscopic analysis of NPs.

Table 1. Particle size, zeta potential, and encapsulation efficiency of NPs are shown. Data represented as mean ± SD (n = 5).
The zeta potential was another parameter determined as a function of surface characteristic of developed formulations (). The zeta potential of GEM-NPs particles was found to be 29.3 ± 1.91 mV, whereas PEG-GEM-NPs and FA-PEG-GEM-NPs show 25.1 ± 1.8 mV and 21.1 ± 1.18 mV, respectively ().
The % drug encapsulation efficiency of GEM-NPs, PEG-GEM-NPs, and FA-PEG-GEM-NPs was found to be 40.8 ± 1.5, 37.2 ± 2.2%, and 39.6 ± 2.7%, respectively. Slightly lesser drug entrapment of PEG-GEM-NPs and FA-PEG-GEM-NPs might possibly be because of the dissolution and subsequent loss of drug adsorbed on the surface, while surface modification in conjugation media was used for PEG and FA-PEG modification. These results are in agreement with the previous findings (Garg et al. Citation2012).
Drug release study
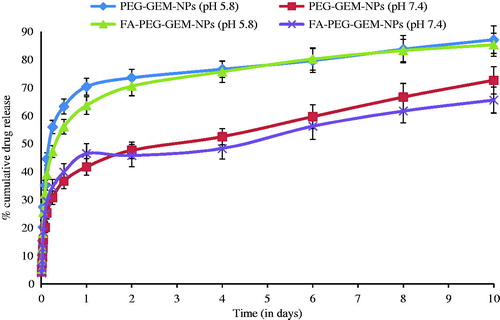
The In vitro drug release of the formulations was studied in PBS (pH= 7.4 and pH= 5.8). The drug release was observed up to 10 d in the case of CS NPs. The total amount of GEM released from the nanoparticles over 10 d was nearly 87% (PEG-GEM-NPs) and 85% (FA-PEG-GEM-NPs) at pH 5.8 and nearly 79% (PEG-GEM-NPs) and 75.3% (FA-PEG-GEM-NPs) at pH 7.4. This observation is indicative of PEG-GEM-NPs’ and FA-PEG-GEM-NPs’ particles that exhibit a biphasic drug-release profile () (Ko et al. Citation2002, Wilson et al. Citation2010), which is characterized as an initial fast drug release which followed slow and sustained release up to 10 d at both pHs (7.4 and 5.8). It was apparent that GEM release rate from NPs at pH 5.8 was rapid compared with that seen with pH 7.4 (). This faster release at lower pH (PBS, pH 5.8) was related to higher solubility of CS at acidic pH, and thereby allowing GEM to leach out at a much faster rate. Hence, FA-PEG-GEM-NPs and PEG-GEM-NPs could be expected to provide a pH-responsive release profile for entrapped GEM in vivo. It might, therefore, help delivering higher drug concentrations into intracellular and tumor interstitium.
Figure 2. In vitro release from the NPs, % cumulative GEM release versus time in PBS (pH 7.4) and PBS (pH 5.8). The data represent the mean ± SD (n = 6).
Cell uptake study
The cell uptake of GEM is a critical step in ensuring the cytotoxic efficiency of drug as it acts by the incorporation of its active triphosphate form (5′-triphosphate GEM) into DNA strand of cell, halting DNA elongation and inducing cell death. The cellular uptake of NPs was determined on A549 cancer cells (. The cellular uptake was seen to show time dependence with all formulations. Cell uptake was found to be increased until 2 h which then followed a not so significant (P > 0.05) trend of cell uptake. This might be probably due to saturation of cells (. The uptake of FITC-labeled FA-PEG-coated NPs (FA-PEG-FITC-NPs) was found significantly greater as compared with that seen with FITC-labeled plain NPs (PEG-FITC-NPs) and plain FITC (. Thus, higher cellular binding with eventual uptake observed with FA-PEG-GEM-NPs is presumably due to greater intracellular delivery of FA-PEG-GEM-NPs by receptor-mediated endocytosis. However, the cellular uptake of FA-PEG-GEM-NPs was significantly (P < 0.001) inhibited by pre-treatment with folic acid. On the contrary, pre-treatment of folic acid with FA-PEG-GEM-NPs leads to preferential binding of folic acid with folate receptors thereby saturating them and ruling out the entry of FA-PEG-GEM-NPs through receptor-mediated endocytosis. The reduced cellular entry upon addition of folic acid and also in the case of uncoated NPs clearly showed that amplified uptake is facilitated by receptors-mediated uptake of FA-PEG-GEM-NPs. Further, cellular uptake of FA-PEG-GEM-NPs is comparable with the cellular uptake of PEG-GEM-NPs. In conclusion, our results are suggestive of the fact that enhanced cellular uptake of FA-PEG-GEM-NPs was specifically mediated by the folic acid receptors.
Figure 3. Graph showing % fluorescent cells after 3 h incubation with PEG-FITC-NPs and FA-PEG-FITC-NPs. The data represent mean ± SD (n = 6).
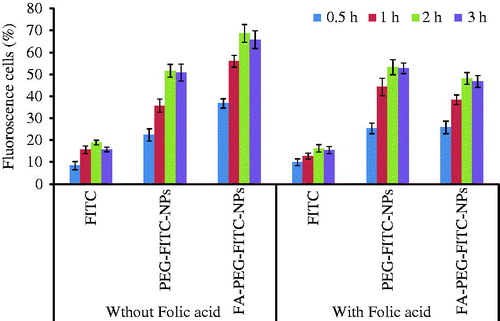
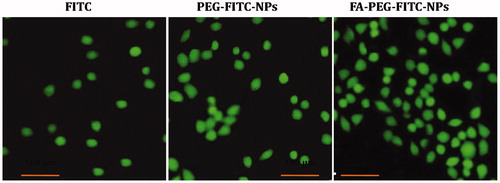
Figure 4. The fluorescence microscopy photomicrographs showing uptake of dye (FITC)-loaded NPs (FITC-NPs) in A549 cell line. The FITC-labeled FA-PEG-FITC-FANPs showed greater uptake in comparison with free FITC, PEG-FITC-NPs.
Cytotoxicity study
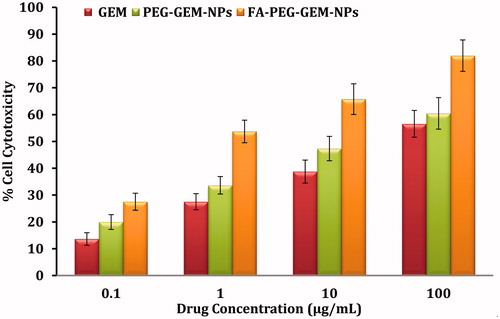
For comparative cytotoxic response, the MTT assay was conducted to investigate the effectiveness of plain GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs (. The results indicate that FA-PEG-GEM-NPs have shown more cytotoxicity in comparison with PEG-GEM-NPs and plain GEM at each and every concentration tested. This is possibly due to receptor–ligand interaction between folic acid receptors (on cell surface) and FA ligand (on NPs surface), which resulted in better interaction between cell and FA-PEG-GEM-NPs. Subsequently, efficient internalization resulted into greater cytotoxic response. The in vitro cell uptake studies also support toxicity data because the presence of FA on the surface of PEG-GEM-NPs significantly drives increased cell uptake when compared with NPs (PEG-GEM-NPs) without FA. It is, therefore, believed that FA plays an instrumental role in the uptake of NPs, and FA-modified PEG-GEM-NPs showed significantly (P < 0.001) better affinity towards A549 cells as compared with PEG-GEM-NPs. The FA receptors lead to an increased uptake of FA-PEG-GEM-NPs, and, therefore, render higher cytotoxic responses (Zhang et al. Citation2015).
Figure 5. The toxicity of different concentrations of plain GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs was determined when these formulations were incubated with A549 cell for 48 h. The data represent mean ± SD (n = 6) different formulations.
In vivo animal studies
Pharmacokinetic studies
The in vivo study suggests the importance of NPs because of alteration in pharmacokinetic and bio-distribution of GEM when delivered through this versatile nanocarrier. The plasma profile of drug () showed that free drug was rapidly (12 h) cleared from blood circulation. On the contrary, GEM delivered through NPs was retained for a significant period of time, i.e. up to as many as 4 d (). Thus, prepared formulation provided slower and sustained drug release for extended periods. In particular, the half-life (t1/2) of GEM was increased from 0.45 ± 0.04 h (free drug) to that of 3.89 ± 0.13 h and 4.05 ± 0.23 h, respectively, for PEG-GEM-NPs and FA-PEG-GEM-NPs (). The free GEM was not detectable 12 h post-administration, whereas GEM shielded with PEG-GEM-NPs and FA-PEG-GEM-NPs was found up to 12 h. These findings are suggestive of the ability of these NPs to extend half-life of said drug. In addition, other pharmacokinetic parameters, i.e. maximum concentration of drug in plasma (Cmax), mean resident time (MRT; h), and the area-under-the-curve (AUC0−t) (), prove the utility of this carrier in drug delivery. The drug confined and retained in the systemic circulation eventually decreases the amount of this anti-tumor agent that was removed from blood stream. The data obtained from pharmacokinetic studies show higher serum concentration of GEM when delivered through FA-PEG-GEM-NPs in comparison with that seen with GEM loaded onto PEG-GEM-NPs. The difference observed in drug concentration of serum profile after PEG-GEM-NPs and FA-PEG-GEM-NPs treatment is closely associated with the differences in surface characteristics, selectivity, and conferred different shielding effects. Therefore, the in vivo bio-distribution study was conducted with an aim to accomplish the fate of nanoparticles in the body.
Figure 6. Plasma profiles of free drug, PEG-GEM-NPs, and FA-PEG-GEM-NPs at different time intervals. The data represent mean ± SD (n = 3).
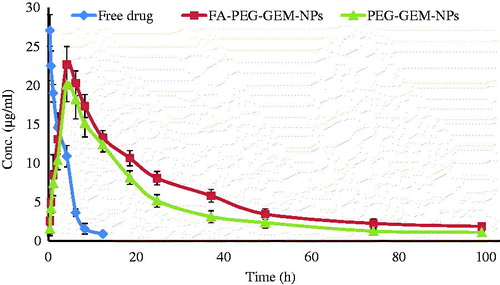
Table 2. Pharmacokinetic parameters in serum of Balb/c mice time. The data represented as mean ± SD (n = 3).
Tissue distribution study
The sustainability and increased residence time of drug in different deep-seated organs was determined by the delivery of GEM through PEG-GEM-NPs and FA-PEG-GEM-NPs (. The greater quantity of GEM was quantified in tumor tissues followed by liver and kidney when delivered through PEG-GEM-NPs’ and FA-PEG-GEM-NPs’ formulations. The greater free drug concentration with FA-PEG-GEM-NPs was estimated in tumor, whereas up to 20.96 ± 2.23% drug was localized after 2 h (), which showed an increase up to 31.33 ± 1.73% after 8 h. The GEM concentration in tumor tissues was estimated to be 13.67 ± 1.95 and 11.67 ± 1.73 with PEG-GEM-NPs’ formulations. These results are indicative of increased targeting efficiency of formulated surface-engineered NPs. The concentration of drug seen in the liver and kidney is comparatively lesser, and almost negligible spleen and heart to render any cell toxicity.
Figure 7. The bio-distribution of free GEM, GEM-loaded onto PEG-GEM-NPs, and FA-PEG-GEM-NPs in different deep-seated tissues at different time intervals. The data represent mean ± SD (n = 3).
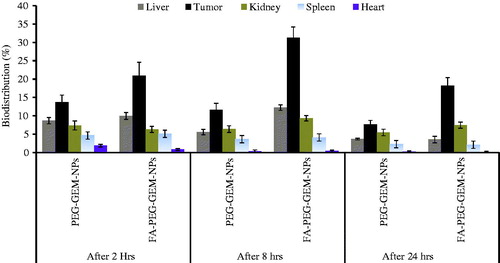
The drug (GEM) content was quantified higher in kidney and, being an important processing organ for clearance of drug, may be accredited to kidney. We saw a steep decrease in free drug concentration when compared with the GEM loaded in PEG-GEM-NPs and FA-PEG-GEM-NPs. The significant decrease in the delivery of GEM loaded onto FA-PEG-GEM-NPs to heart is suggestive of reduced or no cardio-toxicity due to GEM treatment. Furthermore, preferential localization of FA-PEG-GEM-NPs in tumors leads to restricted entry of drug in non-targeted organs. Therefore, low levels of GEM when delivered through folic acid-coated NPs in comparison with PEG-GEM-NPs were observed in tumor tissues. Our results show that FA-PEG-GEM-NPs allowed accumulation of greater amount of drug in tumor as compared with non-targeted organs (. On the contrary, ligand-unanchored NPs show normal distribution of drug among different organs. Consistent to what was reported earlier (Bareford and Swaan Citation2007, Cho et al. Citation2007, Gref et al. Citation1994), different variables such as particle size, polymer composition, molecular weight, and surface characteristic of NPs determine the particle distribution in the body. We confirm and validate the importance of smaller particle size of PEG-GEM-NPs and FA-PEG-GEM-NPs () which is shown to increase residence time of NPs in the body. We report here, as others (Garg et al. Citation2012), that FA plays a key role in selective localization of FA-PEG-GEM-NPs in the lung cancer cells. The FA-anchored PEG-GEM-NPs could be recognized by the folic acid receptors, and eventually transported to lung cancerous cells via receptor-mediated endocytosis. The unmodified PEG-GEM-NPs get accumulated sparely in tumor than FA-PEG-GEM-NPs. The residual PEG-GEM-NPs seen in various organs were due to non-specific binding and uptake by cells of reticulo-endothelial system (RES) (Banerjee et al. Citation2002).
In vivo anti-tumor activity
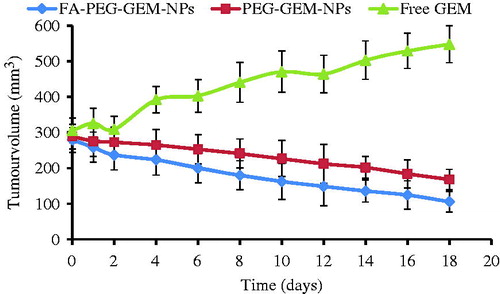
We next decided to assess anti-tumor activity of plain GEM, GEM-loaded PEG-GEM-NPs, and FA-PEG-GEM-NPs (). We saw that free drug failed to reduce the tumor burden/volume. It may be probably due to its rapid clearance from circulation or its less tumor-targeting efficiency. However, free drug, to an extent, showed to put a tab on rapid increase in tumor volume as is evident by constant rise in the curve of the animals treated with normal saline. The GEM-loaded CS NPs were shown to reduce the tumor volume. Interestingly, free drug showed an improved and significantly higher (p < 0.001) anti-tumor activity of GEM delivered through PEG-GEM-NPs and FA-PEG-GEM-NPs at all time points from second day onwards (. It is believed that incorporation of GEM into NPs will shield drug from being metabolized in circulation, and sustained release from NPs matrix might may have improved the anti-cancer activity. The greater reduction in tumor volume was estimated with FA-PEG-GEM-NPs’ and PEG-GEM-NPs’ formulations (. This can be attributed to the active targeting of folic acid-conjugated PEG-GEM-NPs towards the FA receptors overly expressed on tumor surface. Also, it leads to better uptake of NPs than that seen with unconjugated NPs. This may be a characteristic feature of selective accumulation ( and ) of FA-PEG-GEM-NPs in tumor followed by receptor-mediated endocytosis which eventually lead to improved anti-tumor activity when compared with PEG-GEM-NPs. Therefore, optimum therapeutic responses, which improved therapeutic efficacy with minimum adverse effects, were achieved.
Figure 8. Assessment of anti-tumor activity in tumor-bearing animal model by the estimation of tumor burden. The anti-tumor activity of free GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs on days, 3, 7, and 11. The data represent mean ± SD (n = 3).
Toxicity profiling of GEM
The serum levels of urea/creatinine, ALP, ALT, and AST are the biomarkers for renal and liver functioning (). We, based on our findings, report that GEM loaded onto folic acid-engineered PEG-GEM-NPs and PEG-GEM-NPs renders minimal alterations in serum kidney (creatinine and urea) and liver (ALT/AST/ALP) functioning markers in comparison with plain GEM. It suggests that liver and kidney were exposed to free GEM without any shielding rendered to the non-target cells, thereby leading cytotoxic actions. On the contrary, FA-PEG-GEM-NPs and PEG-GEM-NPs due to their sustained release behavior and shielding effect could significantly reduce hepato- and nephro-toxicity. We have seen that kidney and liver functioning parameters were observed slightly higher (P > 0.05) with FA-PEG-GEM-NPs as compared with PEG-GEM-NPs. It is explained based on the presence of folic acid receptor present in liver and kidney (Chen et al. Citation2015).
Table 3. The toxicity profile of plane GEM, PEG-GEM-NPs, and FA-PEG-GEM-NPs. The data represented as mean ± SD (n = 3).
Conclusion and future perspectives
The lung-targeted drug delivery NPs (FA-PEG-GEM-NPs) consisting FA-PEG and CS was prepared and characterized. The presents study indicates the targeting prospective, spatial delivery, amplified bioavailab1ility, and elevated retention potential of the formulation in tumor tissues. Further, the presence of folic acid receptors provided a platform and manifold replica of ligand to facilitate the tumor targeting. The drug delivered through novel NP gets accumulated and maintained in tumor tissues. This accumulation is remarkably higher in ligand-anchored NPs as compared with the plane drug. Further, in vitro cell uptake results showed that the introduction of FA to PEG-GEM-NPs could significantly increase the affinity of particles, and the content therein to human lung carcinoma cells. Our study carried out with GEM-loaded onto FA-PEG-GEM-NPs showed marked cytotoxicity in A549 cells, and complemented by the A549 cell-bearing mice. Finally, the present investigation shows prospective of folic acid-decorated PEG-GEM-NPs as an efficient vector to ferry large doses of anti-cancer drug(s). In conclusion, developed FA-PEG-GEM-NPs nano-particulate system demonstrated minimal toxicity in the area tested. Our observations are indicative of facilitated targeted delivery of anti-cancer drug to tumor sites, with reduced access to non-tumor tissues when surface engineering was done.
Disclosure statement
The authors report no conflicts of interest.
References
- Banerjee T, Mitra S, Kumar Singh A, Kumar Sharma R, Maitra A. 2002. Preparation, characterization and biodistribution of ultrafine chitosan nanoparticles. Int J Pharm. 243:93–105.
- Bareford LM, Swaan PW. 2007. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 59:748–758.
- Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. 2014. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 66:2–25.
- Chen H, Xie LQ, Qin J, Jia Y, Cai X, Nan W, et al. 2015. Surface modification of PLGA nanoparticles with biotinylated chitosan for the sustained in vitro release and the enhanced cytotoxicity of epirubicin. Colloids Surf B Biointerfaces 138:1–9.
- Chen J, Huang L, Lai H, Lu C, Fang M, Zhang Q, Luo X. 2014. Methotrexate-loaded PEGylated chitosan nanoparticles: synthesis, characterization, and in vitro and in vivo antitumoral activity. Mol Pharm. 11:2213–2223.
- Cho YW, Park SA, Han TH, Son DH, Park JS, Oh SJ, et al. 2007. In vivo tumor targeting and radionuclide imaging with self-assembled nanoparticles: mechanisms, key factors, and their implications. Biomaterials 28:1236–1247.
- Fruh M, Gillessen S, Cerny T, Demmer R, D'Addario G. 2008. Two-weekly gemcitabine fixed dose rate and oxaliplatin combination chemotherapy for advanced non-small-cell lung cancer. Lung Cancer 62:344–350.
- Fukuda MN, Ohyama C, Lowitz K, Matsuo O, Pasqualini R, Ruoslahti E, Fukuda M. 2000. A peptide mimic of E-selectin ligand inhibits sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res. 60:450–456.
- Garg NK, Dwivedi P, Campbell C, Tyagi RK. 2012. Site specific/targeted delivery of gemcitabine through anisamide anchored chitosan/poly ethylene glycol nanoparticles: an improved understanding of lung cancer therapeutic intervention. Eur J Pharm Sci. 47:1006–1014.
- Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. 1994. Biodegradable long-circulating polymeric nanospheres. Science 263:1600–1603.
- Hsu C, Kuo SH, Hu FC, Cheng AL, Shih JY, Yu CJ, et al. 2008. Gemcitabine plus conventional-dose epirubicin versus gemcitabine plus cisplatin as first-line chemotherapy for stage IIIB/IV non-small cell lung carcinoma-a randomized phase II trial. Lung Cancer 62:334–343.
- Jain A, Agarwal A, Majumder S, Lariya N, Khaya A, Agrawal H, Majumdar S, Agrawal GP. 2010. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J Control Release 148:359–367.
- Jain A, Jain A, Garg NK, Tyagi RK, Singh B, Katare OP, Webstere TJ, Soni V. 2015. Surface engineered polymeric nanocarriers mediate the delivery of transferrin–methotrexate conjugates for an improved understanding of brain cancer. ActaBiomater 24:140–151.
- Jain A, Kesharwani P, Garg NK, Jain A, Jain SA, Jain AK, et al. 2015. Galactose engineered solid lipid nanoparticles for targeted delivery of doxorubicin. Colloids and Surf B: Biointerfaces 134:027
- Jain S, Mathur R, Das M, Swarnakar NK, Mishra AK. 2011. Synthesis, pharmacoscintigraphic evaluation and antitumor efficacy of methotrexate-loaded, folate-conjugated, stealth albumin nanoparticles. Nanomedicine 6:1733–1754.
- Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. 2004. Cancer statistics, 2004. CA Cancer J Clin. 54:8–29.
- Ji J, Zuo P, Wang YL. 2015. Enhanced antiproliferative effect of carboplatin in cervical cancer cells utilizing folate-grafted polymeric nanoparticles. Nanoscale 10:453.
- Kerr KM. 2001. Pulmonary preinvasive neoplasia. J Clin Pathol. 54:257–271.
- Kesharwani P, Iyer AK. 2015. Recent advances in dendrimer-based nanovectors for tumor-targeted drug and gene delivery. Drug Discov Today 20:536–547.
- Ko JA, Park HJ, Hwang SJ, Park JB, Lee JS. 2002. Preparation and characterization of chitosan microparticles intended for controlled drug delivery. Int J Pharm. 249:165–174.
- Koo OM, Rubinstein I, Onyuksel H. 2005. Role of nanotechnology in targeted drug delivery and imaging: a concise review. Nanomedicine 1:193–212.
- Kumar G, Sharma S, Shafiq N, Khuller GK, Malhotra S. 2012. Optimization, in vitro-in vivo evaluation, and short-term tolerability of novel levofloxacin-loaded PLGA nanoparticle formulation. J Pharm Sci. 101:2165–2176.
- Lee KD, Choi SH, Kim DH, Lee HY, Choi KC.2015. Self-organized nanoparticles based on chitosan–folic acid and dextran succinate-doxorubicin conjugates for drug targeting. Arch Pharm Res. 37:1546–1553.
- Mitra S, Gaur U, Ghosh PC, Maitra AN. 2001. Tumour targeted delivery of encapsulated dextran-doxorubicin conjugate using chitosan nanoparticles as carrier. J Control Release 74:317–323.
- Moghimi SM, Hunter AC, Murray JC. 2001. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 53:283–318.
- Na K, Bum Lee T, Park KH, Shin EK, Lee YB, Choi HK. 2003. Self-assembled nanoparticles of hydrophobically-modified polysaccharide bearing vitamin H as a targeted anti-cancer drug delivery system. Eur J Pharm Sci. 18:165–173.
- Nandini PT, Doijad RC, Shivakumar HN, Dandagi PM. 2015. Formulation and evaluation of gemcitabine-loaded solid lipid nanoparticles. Drug Deliv. 22:647–651.
- O'Shannessy DJ, Yu G, Smale R, Fu YS, Singhal S, Thiel RP, Somers EB, Vachani A. 2012. Folate receptor alpha expression in lung cancer: diagnostic and prognostic significance. Oncotarget 3:414–425.
- Rampino A, Borgogna M, Blasi P, Bellich B, Cesaro A. 2013. Chitosan nanoparticles: preparation, size evolution and stability. Int J Pharm. 455:219–228.
- Sandler AB, Nemunaitis J, Denham C, von Pawel J, Cormier Y, Gatzemeier U, et al. 2000. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 18:122–130.
- Singh R, Shakya AK, Naik R, Shalan N. 2015. Stability-indicating HPLC determination of gemcitabine in pharmaceutical formulations. Int J Anal Chem. 2015: Article ID: 862592. doi:10.1155/2015/862592.
- Tseng CL, Wang TW, Dong GC, Yueh-Hsiu WS, Young TH, Shieh MJ, Lou PJ, Lin FH. 2007. Development of gelatin nanoparticles with biotinylated EGF conjugation for lung cancer targeting. Biomaterials 28:3996–4005.
- Wang M, Hu H, Sun Y, Qiu L, Zhang J, Guan G, et al. 2013. A pH-sensitive gene delivery system based on folic acid-PEG-chitosan – PAMAM-plasmid DNA complexes for cancer cell targeting. Biomaterials 34:10120–10132.
- Wilson B, Samanta MK, Santhi K, Kumar KP, Ramasamy M, Suresh B. 2010. Chitosan nanoparticles as a new delivery system for the anti-Alzheimer drug tacrine. Nanomedicine 6:144–152.
- Yu X, Yang G, Shi Y, Su C, Liu M, Feng B, Zhao L. 2015. Intracellular targeted co-delivery of shMDR1 and gefitinib with chitosan nanoparticles for overcoming multidrug resistance. Int J Nanomed. 10:7045–7056.
- Zhang BF, Xing L, Cui PF, Wang FZ, Xie RL, Zhang JL, et al. 2015. Mitochondria apoptosis pathway synergistically activated by hierarchical targeted nanoparticles co-delivering siRNA and lonidamine. Biomaterials 61:178–189.
- Zwicke GL, Mansoori GA, Jeffery CJ. 2012. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 3:1–11.