
Abstract
Hepatocellular carcinoma (HCC) shows low response to most conventional treatment strategies. Therefore, there is an urgent need for new and effective chemotherapies. Nanotechnology gives a dramatic impact on medicine. In this work, paclitaxel loaded nanoparticles (NPs) decorated with bivalent fragment HAb18 F(ab’)2 and/or cell penetrating peptide (CPP) were developed and evaluated. NPs were prepared by emulsification-solvent evaporation method and decorated by carbodiimide chemistry. The physicochemical characteristics of NPs (i.e. encapsulation efficiency, particle size distribution, morphology, release in vitro) were investigated. Cellular uptake and accumulation in tumor tissue of NPs were determined. To assess anti-tumor activity of NPs in vitro and in vivo, cell survival assay and tumor regression study were carried out using HCC cell lines (HepG2 and Huh7) and their xenografts. Average particle size of all NPs was between 100 and 200 nm. Drug-loaded NPs possessed spherical morphology and higher encapsulation efficiency. The accumulation of NPs decorated with HAb18 F(ab’)2 and CPP depended on dual effects of passive and active targeting. Drug loaded nanoparticles showed cytotoxicity on the tumor cells in vitro and in vivo. NPs decorated with HAb18 F(ab’)2 and CPP showed maximization of therapeutic action for targeting and effective endocytosis. These results suggest that the nano-drug delivery system could be a promising candidate with excellent therapeutic efficacy for HCC therapy.
Introduction
Hepatocellular carcinoma (HCC) is a malignant tumor with high morbidity and mortality [Citation1]. Although several therapeutic approaches can be used for the treatment of HCC, including surgery, radiofrequency ablation (RFA), percutaneous ethanol injection (PEI) and chemoembolization (TACE), existing therapies are limited in the ability to cure the malignancy and to prevent its metastases and relapses [Citation2]. Therefore, for the larger part of HCC, chemotherapy remains to be the alternative effective treatment strategy in tumor suppressor and elimination [Citation3].
Biodegradable polymeric nanoparticles (NPs) have emerged as one of the most promising delivery and targeting carrier systems for many drugs relative to free drugs [Citation4,Citation5]. Ideal NPs can minimize drug degradation and inactivation upon administration, prevent undesirable side effects and increase drug bioavailability and the fraction of drug delivered in the pathological area. The small size makes NPs to escape from renal exclusion and the reticuloendothelial system (RES) and gives them an enhanced permeation and retention (EPR) effect via tumor vessels, termed the passive targeting. For active targeting, the strategy is to decorate carriers with the ligands, such as antibody [Citation6–8].
Paclitaxel is a highly potent anticancer drug for different cancers, especially cervical, breast, lung and ovarian cancers [Citation9]. There are many reports in the literature regarding the activity of paclitaxel against HCC in vitro and in vivo [Citation10–12]. Due to poor water solubility, paclitaxel is traditionally intravenously administered with cremophor EL® [polyethoxylated castor oil and ethanol (1:1, v/v)] as a solvent. However, cremophor EL may cause serious undesired side-effects and lead to hypersensitivity reactions in many patients. Recently, an albumin-bound paclitaxel NPs (Abraxane™) is the first Cremophor EL-free paclitaxel agent which was approved for clinical application by the Food and Drug Administration [Citation13]. Our previous study suggested the selective therapeutic effect of targeted paclitaxel loaded NPs on liver cancer stem cells [Citation14].
Bivalent fragment HAb18 F(ab’)2 is a specific cancer-targeting antibody that recognizes and interact with HAb18G overexpressed in HCC [Citation15]. Our previous work reported that DOX–PLGA–PEG micelles decorated with HAb18 F(ab’)2 resulted in significant improvement in therapeutic response on HCC in vitro and in vivo [Citation16].
Cell penetrating peptide (CPP) is defined as the peptide sequence of less than 50 amino acids capable of transporting various molecules efficiently into cells across membranes. CPP has become a powerful tool for the intracellular delivery of various agents that are inherently almost impermeable, including peptides and proteins, nucleic acids, drugs and NPs [Citation17,Citation18]. Polyarginine, RRRRRRRRR (R9), is one of the most commonly used CPP’s [Citation19]. Combination of antibody and CPP’s in NPs can provide chemotherapy with cell specificity and enhanced uptake [Citation20].
In this work, paclitaxel-loaded NPs decorated with bivalent fragment HAb18 F(ab’)2 and polyarginine were developed and evaluated to improve HCC chemotherapy.
Materials and methods
Materials
Reagents
99.7% purity paclitaxel was from Beijing HuaFeng United Technology (Beijing, China). PLGA (L/G = 50/50, Mw = 25,000) was from Chengdu Institute of Organic Chemistry, Chinese Academy of Science (China). The bivalent fragment HAb18 F(ab’)2 was from Cell Engineering Research Center, Fourth Military Medical University. Fluorescein isothiocyanate (FITC), propidium iodide (PI), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), bovine serum albumin (BSA), Dichloromethane (DCM) and Coomassie brilliant blue G-250 and polyvinyl alcohol (PVA, Mw = 30,000–70,000) were from Sigma-Aldrich (St. Louis, MO). RRRRRRRRR and fluorescent GLPK (FITC) RRRRRRRRR were synthesized by Shanghai GL Biochem Co. Ltd (Shanghai, China).
Cells
Human HCC cell lines HepG2 and Huh7 cells were obtained from Laboratory of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University. The cells were maintained in monolayers in DMEM medium supplemented with fetal bovine serum (10%), penicillin (100 units/ml) and streptomycin (50 units/ml) at 37 °C in a humidified incubator with 5% CO2 atmosphere. For experiments, cells in exponential growth phase were used.
Animals
Male BALB/c nude mice (5–6 weeks old) were purchased from the Experimental Animal Center of Fourth Military Medical University. All animals were maintained in autoclaved microisolator cages housed in a positive pressure containment rack. Mice received care in accordance with the guidance suggestions for the Care and Use of Laboratory Animals. The study protocol of animals care and procedures were approved by the ethics committee of the hospital of Xidian group.
Methods
Preparation of NPs
Blank PLGA NPs without drug (B-NPs) and paclitaxel loaded PLGA NPs (P-NPs) were prepared by oil-in-water emulsification solvent evaporation method [Citation16]. Fluorescent NPs were prepared adding 10 mg of FITC (0.25% (w/v)) [Citation17,Citation18]. The incorporated dye, as a probe for NPs, offers a sensitive method to determine their intracellular uptake and retention.
Conjugation of ligands to NPs
HAb18 F(ab’)2 was diluted in pH 5.7 PBS at a concentration of 1 mg/ml. Polyarginine was diluted in pH 7.4 PBS at a concentration of 2 mg/ml. For covalent conjugation of HAb18 F(ab’)2 antibody fragment, polyarginine (fluorescent polyarginine used for calculation of loading efficiency) and the combination on P-NPs (H-NPs, C-NPs and H + C-NPs), EDC/NHS chemistry was employed [Citation18]. The supernatant was collected for estimation of unconjugated antibody fragment and/or polyarginine. The NPs decorated with BSA (BSA-NPs) were prepared by the same method as protein-conjugated control. Total amount of antibody and polyarginine in the supernatant was determined by Coomassie brilliant blue method. Amount of polyarginine was determined using fluorescence spectrophotometry. Loading efficiency = amount of (antibody or polyarginine) in NPs/amount of NPs.
Drug content
Paclitaxel containing in NPs was determined by high-performance liquid chromatography (HPLC) system [Citation21]. A calibration curve was obtained using a series of paclitaxel solutions at different concentrations. Loading efficiency = amount of drug in NPs/amount of NPs. Encapsulation efficiency = amount of drug in NPs/initial amount of drug.
Particle size and morphology
The mean diameter of NPs was determined by laser light scattering with the ZetaSizer NanoZS analyzer (Malvern Instruments, Malvern, UK). The surface morphology of NPs was observed.
Examination of drug release
5 mg of NPs was suspended in phosphate buffer saline (PBS, pH 7.4) and continually shaken at 120 rpm at 37 °C. The supernatant was taken out at different time points, meanwhile the same volume of fresh PBS was added to replace the supernatant. The concentration of paclitaxel in all samples was determined by HPLC analysis.
Cellular uptake
HepG2 and Huh7 cells were seeded on coverslips in a six well tissue culture plate. Fluorescent H + C-NPs at a concentration of 1 mg/ml were added and was incubated for 12 h. After washing with PBS, the cells were stained using PI (3 μg/ml) and photographed by fluorescence microscope. FITC and PI showed green and orange, respectively. H + C-NPs on surface of cells were observed by SEM. After incubation with H + C-NPs, the cells were harvested by trypsinization and fixed with glutaraldehyde. Internalization of NPs was further measured by transmission electron microscopy (TEM, JEOL, Japan).
Flow cytometry
HepG2 and Huh7 cells were incubated with 1 mg/ml fluorescent P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs in culture flasks for 12 h. The cells were collected, washed and resuspended in PBS. Cellular fluorescence intensity was analyzed by flow cytometry (Beckman-Coulter, USA).
Cell morphology
HepG2 and Huh7 cells were incubated for 24 h with 1 mg/ml of P-NPs, H-NPs, C-NPs and H + C-NPs (54.24, 54.03, 53.85 and 53.67 ng/ml paclitaxel released). Cell morphology was recorded by phase-contrast photomicrography.
Cell survival assay
HepG2 and Huh7 cells were plated at 500 cells per well in six well tissue culture plates for 1, 2, 4, 8, 12 and 24 h with 1 mg/ml of B-NPs, P-NPs (6.95, 10.82, 15.77, 22.46, 35.43 and 54.24 ng/ml paclitaxel released), BSA-NPs (6.81, 10.21, 14.95, 21.78, 34.77 and 52.15 ng/ml paclitaxel released), H-NPs (7.01, 10.28, 15.29, 21.85, 34.92 and 54.03 ng/ml paclitaxel released), C-NPs (6.83, 10.21, 14.79, 22.08, 35.11 and 53.85 ng/ml paclitaxel released) and H + C-NPs (6.77, 9.68, 14.56, 21.87, 34.41 and 53.67 ng/ml paclitaxel released), respectively. PBS was added as a control. After administration, the cells were continued to culture for 14 days. The macroscopic colonies were stained with Giemsa and counted manually. Cell viability was measured by the ability of single cell to form colonies in vitro.
Bio distribution
To establish human HCC xenografts model, HepG2 cells (1 × 106) were inoculated by subcutaneous injection into the back of nude mice for 14 days. For administration, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs were suspended in a certain volume of PBS in order to obtain the required concentration. The formulation was injected through the tail vein at the dose of 1 mg/kg of mouse. After 2 h, mice were sacrificed by cervical dislocation and tissues (tumor, brain, heart, lung, liver, kidney and spleen) and blood were obtained. 500 mg of tissues was homogenated and blood plasma was prepared. Drug in homogenate and blood plasma was determined using HPLC.
Anti-tumor activity in vivo
Mice bearing HepG2 tumors were randomly divided into 7 groups (n = 6). Mice were injected via the tail vein with NPs at 1 mg/kg/day each third day (six times) for 15 days. Mice as control were administrated with PBS. The tumor size was monitored daily by a fine caliper and the tumor volume was calculated as (major axis) × (minor axis)2 × 1/2. Inhibition rate of tumor = (tumor volume of control - tumor volume of experiment)/tumor volume of control.
Statistical analysis
All data were expressed as the mean ± standard deviation. Statistical analysis was completed using SPSS19.0 (SPSS Inc., Chicago, IL). The One-Way ANOVA was used to determine the statistical differences with the level of significance set at p < .05.
Results and discussion
Characterization of NPs
An EDC/NHS covalent coupling method to modify polymer carriers with ligands is well established for multi-functional NPs. In this study, amino-groups of HAb18 F(ab’)2 and polyarginine were conjugated to carboxylic end groups of PLGA via an amide linkage. Loading efficiency of antibody in H-NPs and H + C-NPs were 1.72 and 1.58%, respectively. Loading efficiency of CPP in C-NPs and H + C-NPs were 3.81 and 3.65%, respectively. The data suggested that these molecules were bound with NPs, but did not indicate whether their biological activity still was preserved.
Particle size plays a key role in tumor accumulation by EPR effect and intracellular transportation. Size of B-NPs, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs were 111.35 ± 47.14, 118.47 ± 52.21, 164.71 ± 59.18, 155.28 ± 68.34, 143.86 ± 65.29 and 175.51 ± 60.31 nm with polydispersity index 0.117, 0.122, 0.143, 0.145, 0.136 and 0.158, respectively (). These particle sizes were suitable for delivery in circulation, because they were large enough to avoid kidney filtration (>10 nm) but also small enough to cross over tissues, to approach cell surface receptors and to aid the endocytosis [Citation21,Citation22]. As conjugating ligands to NPs surface, the increase of particle size was presumable. B-NPs, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs showed the zeta potential of −38.4, −41.6, −30.7, −16.4, −21.3 and −12.6 mV, respectively. The increase in the zeta potential was due to the positive charge of HAb18 F(ab’)2 and polyarginine and their repulsion. SEM observation confirmed that all NPs had smooth surface, sphere-like shape and monodispersed size.
Figure 1. (A) Particle size distribution [green curve (the middle peak): H-NPs, blue curve (the shortest peak): C-NPs, red curve (the highest peak): H + C-NPs)] and (B) SEM photographs of all NPs.
![Figure 1. (A) Particle size distribution [green curve (the middle peak): H-NPs, blue curve (the shortest peak): C-NPs, red curve (the highest peak): H + C-NPs)] and (B) SEM photographs of all NPs.](/cms/asset/525c3858-d4bf-4f33-b168-2e1a5b708878/ianb_a_1360325_f0001_c.jpg)
NPs formulated from PLGA polymer offer a non-toxic and efficient carrier system for drug delivery. The drug encapsulation efficiency is crucial for NPs clinical applications. PLGA polymer of >80% is considered sufficient for preparation of the efficient NPs. Meanwhile, the high loading capacity could reduce the quantity of the carriers for administration. In our work, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs had an encapsulation efficiency of 88.57 ± 9.1%, 86.19 ± 7.7%, 87.04 ± 7.8%, 87.46 ± 6.9%, 85.86 ± 8.5% and a loading efficiency of 4.58 ± 0.56%, 4.47 ± 0.48%, 4.51 ± 0.61%, 4.54 ± 0.49%, 4.45 ± 0.54%, respectively. The encapsulation efficiency of paclitaxel is consistent with previous studies due to its high partition coefficient and retention in the organic phase as solidify [Citation23,Citation24]. The loading efficiency among all of NPs did not show significant difference.
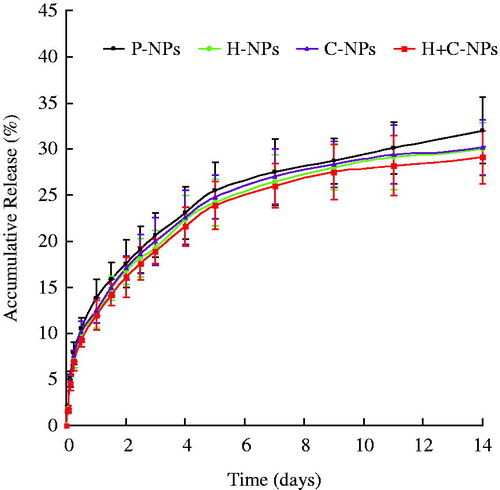
Paclitaxel was released from the polymer matrix in a biphasic pattern: an initial fast release and a following slower and continuous release with no more than 35% of drug released within 14 days. Approximately 15% drug was released on the first day (). The initial burst effect can be ascribed to the release of drug loosely bound on NPs by mechanisms of hydrolysis and diffusion, while the slower and sustained release depended on the water-mediated erosion and degradation of the polymer. The release profile suggested that paclitaxel release from NPs could be controlled by time in the early stage. The kinetic data showed no significant difference among all of NPs. Therefore, the rate and extent of drug release from NPs was not affected by modification of ligands.
Figure 2. The release profiles of P-NPs, H-NPs, C-NPs and H + C-NPs in vitro.
In vitro anti-tumor activity
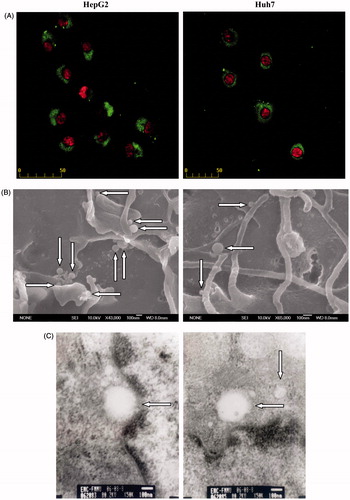
Due to cell membrane barriers and clearance from the body, efficient intracellular delivery of nano-carriers has always been a critical problem. After 12 h of treatment with H + C-NPs, showed internalization of H + C-NPs by HCC cells using fluorescence microscopy. However, SEM indicated that NPs maybe adhered and located on surface of cells. Finally, TEM demonstrated that NPs were predominantly within cells.
Figure 3. Cellular uptake of H + C-NPs. (A) The fluorescence microscopy photographs of HCC cells internalizing nanoparticles, in 400× magnification. (B) The SEM photographs of H + C-NPs on the surface of cells and (C) TEM photographs of H + C-NPs within cells.
In order to assess the ability to penetrate cells quantitatively, NPs were labeled with PI and determined by flow cytometry. HepG2 and Huh7 cells cultured with fluorescent H-NPs, C-NPs and H + C-NPs showed more fluorescence intensity than fluorescent P-NPs and BSA-NPs. There was no difference in fluorescence intensity between fluorescent P-NPs and BSA-NPs. Among fluorescent H-NPs, C-NPs and H + C-NPs, fluorescence intensity of fluorescent H + C-NPs was most significant to show the synergistic effect (). Many studies have demonstrated that NPs enter the cells through specific and nonspecific endocytosis. Our results suggested that specifically antigen-antibody recognition improved uptake of NPs and CPP as a promoting factor enhanced the intracellular delivery.
Figure 4. Intracellular fluorescence intensity of HCC cells internalizing P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs (*p < .05 vs. P-NPs and BSA-NPs; **p < .05 vs. H + C-NPs).
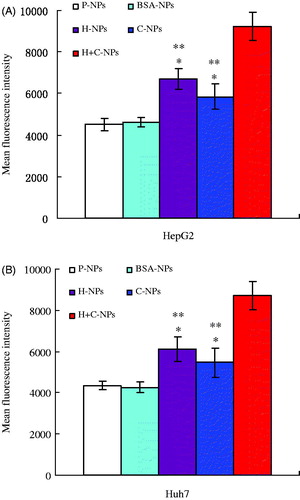
The results also indicated that BSA did not effected internalization of NPs. It was proven indirectly that HAb18 F(ab’)2 and polyarginine kept their biological activity.
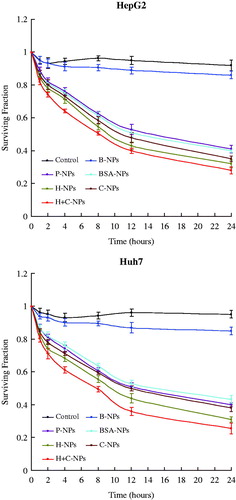
Intracellularly retained NPs could slowly release the encapsulated drug leading to a sustained action. Exposed to drug loaded NPs, the morphological changes of HCC cells are shown in . Paclitaxel released from NPs lead to degenerative and necrotizing injuries of cells. Cell survival assay showed that drug loading NPs significantly inhibited the viability and proliferation of HepG2 and Huh7 cells. The effects of H-NPs, C-NPs and H + C-NPs were more significant than that of P-NPs and BSA-NPs. There was no therapeutic difference between P-NPs and BSA-NPs. H + C-NPs exhibited maximization of therapeutic action among all drug loaded NPs (). This was consistent with data of flow cytometry. The effectiveness of H + C-NPs was due to the following mechanisms: increase of drug concentration near the HCC cells by antigen-antibody specific recognition and improved endocytosis by non-specific CPP pathway.
Figure 5. Phase-contrast photomicrographs of HepG2 and Huh7cells following a 24 h treatment with P-NPs, H-NPs, C-NPs and H + C-NPs, respectively.
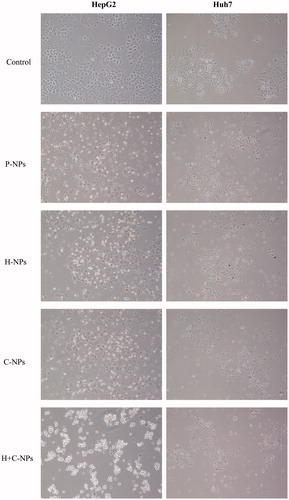
Figure 6. The effects of all NPs on the surviving fractions of HCC cell lines.
In vivo anti-tumor activity
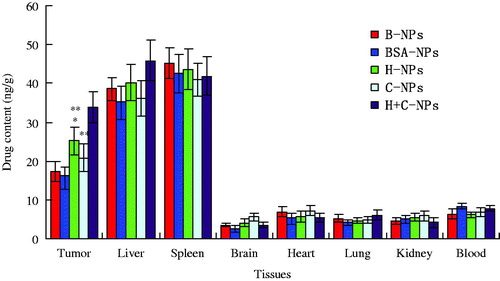
The bio distribution efficiency of paclitaxel in different tissues of mice was shown in . The high amount of drug was detected in tumor, liver and spleen for H-NPs and H + C-NPs, but only in liver and spleen for P-NPs, BSA-NPs and C-NPs. The accumulation of H + C-NPs in tumor was more than that of H-NPs. However, drug in brain, heart, lung, kidney and blood were obviously less or nearly nothing. The bio distribution studies revealed that NPs were located in different organs, regardless of their size, shape and material. Generally, most particulate delivery systems are mainly accumulated in liver and spleen due to nonspecific phagocytosis and elimination by the RES after intravenous injection. Tumor tissues have loose interconnections and focal intercellular openings in size from 100 to 1000 nm between defective endothelial cells [Citation25]. Our results indicated that the average size of all NPs was between 100 and 200 nm. Consequently, this passive targeting can effectively enhance the accumulation at the tumors and alleviate the toxicity to normal tissues. So far the polymeric NPs have mostly lacked active targeting and instead rely mainly on the EPR effect. In order to solve the problem, targeted NPs decorated with ligands may constitute the next generation of polymeric drug delivery systems. First, targeted NPs slowly accumulate in tumor tissue and ultimately reached high levels due to the EPR effect. Second, non-ligand decorated NPs remain in the interstitial space and are subject to decomposition, degradation or phagocytosis with drug release. Ligand decorated NPs bind to and internalize in tumor cells via specific interactions [Citation26]. CPPs have been used to affect the intracellular delivery of many classes of bioactive cargo, including small molecules, nucleic acids, antibodies and NPs [Citation27,Citation28]. This study, has a convincing evidence that CPPs enhanced therapeutic potential of NPs for HCC cells.
Figure 7. The distribution profiles of paclitaxel in different tissues of nude mice bearing HepG2 xenograft (*p < .05 vs. P-NPs and BSA-NPs; **p < .05 vs. H + C-NPs).
In tumor regression study, showed tumor volume progress of HepG2 xenograft for 60 days. Drug loading NPs exhibited therapeutic profiles in terms of tumor growth inhibition and survival rates. NPs decorated with HAb18 F(ab’)2 suppressed tumor growth more markedly than non-targeted NPs. H + C-NPs showed most significant anti-cancer action among all the formulations. Accumulation of targeted NPs in tumor microenvironment depends on dual effects of passive and active targeting. Improved transmembrane delivery of NPs by CPPs offers enormous potential for therapeutic intervention in diseased cells. Enhanced extracellular and intracellular accumulation of H + C-NPs in tumor showed promising efficacy compared with routine NPs. Effect of C-NPs was more obvious than that of P-NPs and BSA-NPs.
Figure 8. (A) In vivo anti-tumor activity assay (the curves of ![]()
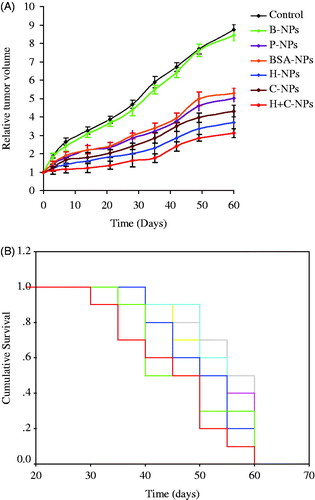
3 days after the final administration, inhibition rates of tumor were 54.8, 52.4, 69.5, 57.9 and 77.3% for P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs, respectively. Tumor volumes between PBS and B-NPs nearly had no variation. Drug loading NPs significantly prolonged the life spans of mice bearing xenograft and improved their survival rates. When observed even up to 60 days, the survival rates were 0, 0, 19.4, 15.7, 27.8, 20.5 and 36.3% for PBS, B-NPs, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs, respectively. The median survival times were 18.6 ± 2.6, 20.4 ± 2.1, 32.2 ± 3.3, 30.5 ± 5.2, 42.4 ± 4.7, 35.2 ± 3.9 and 49.7 ± 5.4 days for PBS, B-NPs, P-NPs, BSA-NPs, H-NPs, C-NPs and H + C-NPs, respectively (). These results suggested that NPs decorated with antibody and/or CPPs might result in a beneficial gain for HCC chemotherapy. With active targeting and effective endocytosis, H + C-NPs showed the maximization of therapeutic response in all NPs. Therefore, this study provides a promising candidate with excellent therapeutic efficacy on HCC. Based on these findings, further studies on administration routes, including via hepatic artery and intratumorally, are significant and encouraged.
Conclusions
Paclitaxel loaded NPs decorated with HAb18 F(ab’)2 and/or polyarginine were developed by EDC/NHS chemistry for targeted delivery and effective endocytosis. Drug loaded NPs showed cytotoxicity on HCC cells in vitro and in vivo. Specificity and higher uptake of H + C-NPs most significantly improved the therapeutic effect. These results suggested that the multifunctional nanosystems with targeting and cell-penetrating abilities could become potential gain for HCC chemotherapy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255.
- Balogh J, Victor III D, Asham EH, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3:41–53
- Colagrande S, Inghilesi AL, Aburas S, et al. Challenges of advanced hepatocellular carcinoma. World J Gastroenterol. 2016;22:7645–7659.
- Kamaly N, Yameen B, Wu J, et al. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116:2602–2663.
- Bogart LK, Pourroy G, Murphy CJ, et al. Nanoparticles for imaging, sensing and therapeutic intervention. ACS Nano. 2014;8:3107–3122
- Allegra A, Penna G, Alonci A, et al. Nanoparticles in oncology: the new theragnostic molecules. Anticancer Agents Med Chem. 2011;11:669–686.
- Chen H, Gao J, Lu Y, et al. Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J Control Release. 2008;128:209–216.
- Liu D, Yang F, Xiong F, et al. The smart drug delivery system and its clinical potential. Theranostics. 2016;6:1306–1323.
- Bernabeu E, Cagel M, Lagomarsino E, et al. Paclitaxel: what has been done and the challenges remain ahead. Int J Pharm. 2017;526:474–495.
- Geng CX, Zeng ZC, Wang JY. Docetaxel inhibits SMMC-7721 human hepatocellular carcinoma cells growth and induces apoptosis. World J Gastroenterol. 2003;9:696–700.
- Jin C, Li H, He Y, et al. Combination chemotherapy of doxorubicin and paclitaxel for hepatocellular carcinoma in vitro and in vivo. J Cancer Res Clin Oncol. 2010;136:267–274.
- Iesalnieks I, Tange S, Scherer MN, et al. Paclitaxel promotes liver graft survival in rats and inhibits hepatocellular carcinoma growth in vitro and is a potentially useful drug for transplant patients with liver cancer. Transplant Proc. 2002;34:2316–2317.
- Ai D, Guan Y, Liu XJ, et al. Clinical comparative investigation of efficacy and toxicity of cisplatin plus gemcitabine or plus abraxane as first-line chemotherapy for stage III/IV non-small-cell lung cancer. Onco Targets Ther. 2016;9:5693–5698.
- Jin C, Yang Z, Yang J, et al. Paclitaxel-loaded nanoparticles decorated with anti-CD133 antibody: a targeted therapy for liver cancer stem cells. J Nanopart Res. 2014;16:2157–2171.
- Xu J, Xu HY, Zhang Q, et al. HAb18G/CD147 functions in invasion and metastasis of hepatocellular carcinoma. Mol Cancer Res. 2007;5:605–614.
- Jin C, Qian N, Zhao W, et al. Improved therapeutic effect of DOX–PLGA–PEG micelles decorated with bivalent fragment HAb18 F(ab’)2 for hepatocellular carcinoma. Biomacromolecules. 2010;11:2422–2431.
- Svensen N, Walton JG, Bradley M. Peptides for cell-selective drug delivery. Trends Pharmacol Sci. 2012;33:186–192.
- Zhang D, Wang J, Xu D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J Control Release. 2016;229:130–139.
- Kanwar JR, Gibbons J, Verma AK, et al. Cell-penetrating properties of the transactivator of transcription and polyarginine (R9) peptides, their conjugative effect on nanoparticles and the prospect of conjugation with arsenic trioxide. Anticancer Drugs. 2012;23:471–482.
- Da Silva CG, Rueda F, Löwik CW, et al. Combinatorial prospects of nano-targeted chemoimmunotherapy. Biomaterials. 2016;83:308–320
- Mcdonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5385.
- Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740.
- Jin C, Bai L, Wu H, et al. Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells. Pharm Res. 2009;26:1776–1784.
- Jin C, Bai L, Wu H, et al. Radiosensitization of paclitaxel, etanidazole and paclitaxel + etanidazole nanoparticles on hypoxic human tumor cells in vitro. Biomaterials. 2007;28:3724–3730.
- Kohn S, Nagy JA, Dvorak HF, et al. Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels. Lab Invest. 1992;67:596–607.
- Wesselinova D. Current major cancer targets for nanoparticle systems. Curr Cancer Drug Targets. 2011;11:164–183.
- Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–393.
- Kristensen M, Nielsen HM. Cell-penetrating peptides as carriers for oral delivery of biopharmaceuticals. Basic Clin Pharmacol Toxicol. 2016;118:99–106.