
Abstract
Tissue engineering strategies have been developed to optimize osseointegration in dental implant surgery. One of the major problems is the non-homogeneous spatial cell distribution in the scaffold, as well as subsequent matrix production. Insufficient nutrient and oxygen supplies inside the scaffold are factors in this phenomenon. To mediate this gradient formation, we have implemented a perfusion culture method to seed human bone marrow mesenchymal stem cells (MSCs) into three-dimensional (3-D)-allogenic bone scaffolds in combination with a marine haemoglobin, HEMOXCell®, for oxygen delivery. Cell culture was performed under static and perfusion conditions, with standard and osteogenic media, with and without HEMOXCell®. The cell seeding efficiency, as well as MSC/scaffold cytocompatibly were assessed using viability and proliferation assays. Scaffolds’ cellularization and extracellular matrix (ECM) formation were analyzed using scanning electron microscopy and histological staining. Cell differentiation was investigated with osteogenic biomarkers gene expression analysis. The perfusion culture was observed to significantly promote MSC proliferation and differentiation throughout the scaffolds, especially when using the induction medium w/HEMOXCell®. Our data suggest that perfusion culture of MSC into allogenic bone substitute with HEMOXCell® as a natural oxygen carrier is promising for tissue engineering applications to oxygenate hypoxic areas and to promote cellular proliferation.
Introduction
In dental surgery, the use of dental implants is a standard procedure for replacing a missing tooth. In a healthy bone, osseointegration is supported by the local inflammation, the neovascularization, the osteogenesis, the bone cells and the bone marrow progenitor cells [Citation1–3]. However, it has been demonstrated that osseointegration is delayed for compromised bone. Indeed, traumatic or pathological injuries, such as diabetes and irradiations, dramatically increased the risks of implant failure due to modifications of the bone microenvironment which impact molecular interactions, cellular functions and thus bone regeneration [Citation4,Citation5]. One of the challenges for prosthodontic rehabilitation in maxillofacial surgeries is to promote implant anchorage into bone which has a poor natural healing capacity. The most common strategy is the replacement of damaged bone using a bone graft substitute. Autologous grafts are considered the “gold standard” treatment due to their osteoconductive, osteoinductive and osteogenic natural properties [Citation6]. However, the availability and morbidity of donor site, and the potential risk of infection represent some of the limitations to this procedure [Citation6]. Allogenic substitutes appear to be an alternative to autogenous bone graft because of their availability without the requirement of a second surgical site [Citation7]. However, the theoretical risks of immune reaction and disease transmission have led to the development of acellular bone substitutes like freeze-dried bone allograft (FDBA) and demineralized freeze-dried bone allografts (DFDBA). These allografts are processed and sterilized using specific protocols to remove biological components including cells and lipids, to minimize the risk of immune reactions and infections of the host [Citation8]. Unfortunately, treatments performed on the collected bone reduce the osteoinductive and osteogenic properties of the graft [Citation9]. As cells and vessels have been eliminated, critical failures of bone substitute grafts can also be attributed to the local ischemia and to the lack of osteoblast progenitor cells into the graft [Citation10].
As a solution, tissue engineering strategies can provide an osteogenic character to an inert graft by creating a new bone-like physicochemical environment using growth factors and osteocompetent cells [Citation11,Citation12]. These strategies should promote healing, decrease the patient’s pain and increase the implant’s biointegration.
Mesenchymal stem cells (MSCs) constitute the most promising candidates for bone tissue engineering because of their immunomodulatory properties and their plasticity. MSCs have been described by Friedenstein as adherent bone marrow cells with a self-renewing capacity, exhibiting a fibroblast-like morphology [Citation13]. MSC can give rise to mesenchymal cell lineages including adipocytes, chondrocytes and osteoblasts in vitro [Citation14]. In addition, MSCs have even been used in the treatment of a large range of tissue repair including cartilage, tendon, nerve, heart and bone [Citation15–17]. MSCs derived from bone marrow are the most frequently used for bone regeneration [Citation18–20]. However, other sources like adipose tissue and peripheral blood have been investigated for bone repair as well [Citation21–23]. MSC can promote new blood vessels formation and limit the immune reaction [Citation24]. Therefore, the use of human platelet lysate (hPL) as a supplement for MSC culture has demonstrated improvements in cellular proliferation, differentiation and angiogenic properties [Citation25,Citation26]. hPL is a rich source of growth factors like platelet-derived growth factor (PDGF), transforming growth factor (TGF), insulin growth factor (IGF), vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) which promote the healing process, angiogenesis and cell osseo-differentiation [Citation25,Citation27–30]. In dental implantology, hPL injections influence the cell migration to the implant and the bone formation [Citation2]. For bone engineering constructs, the technique employed for seeding and the subsequent culture conditions, can affect cell viability as well as their proliferation and plasticity. Perfusion culture system has been reported to promote cell and nutrient repartition as well as oxygen diffusion and cell differentiation to osteogenic lineages [Citation31–33].
HEMOXCell® is a biological oxygen carrier extracted from the marine worm Nereis virens [Citation34,Citation35] and produced by the French biotechnology company HEMARINA in compliance with the good manufacturing practices (GMP) requirements [Citation36]. This extracellular haemoglobin, which can carry 40 times more oxygen molecules than the human haemoglobin, enables oxygen release in a simple oxygen gradient according to cellular needs. We recently reported that HEMOXCell® can be used as an oxygenation additive in CHO-S culture for bioproduction, accelerating the cell productivity as well as the growth rate [Citation37]. In another study, we have highlighted its potential use at low doses as a non-toxic additive for bone marrow MSC culture with positive effects on cell proliferation with a 25% increase cell growth and the maintenance of cell viability and characteristics [Citation38].
Given its potential for oxygenation, we aim to develop a method associating HEMOXCell® and human MSCs, to promote cell growth and differentiation in three-dimensional (3-D) FBDA substitutes (BIOBank®). In order to achieve it, we investigated the cell behaviour in both static and perfused 3-D cultures, using osteoinductive hPL-supplemented medium with HEMOXCell®. We demonstrated that perfusion of bone marrow MSC through the pores of BIOBank® substitutes resulted in the efficient cellular expansion and differentiation into osteoblastic lineage especially in HEMOXCell®-supplemented medium. This work underlines the potential use of a universal oxygen carrier for in vitro bone substitute cellularization as well as providing an experimental basis for the further in vivo studies. Among these, a study on the oxygenation of hypoxic graft areas and environment in the implantation site where the local blood supply is compromised would be of interest.
Materials and methods
HEMOXCell®
The extracellular marine haemoglobin is manufactured under the trademark name of HEMOXCell® or sometimes referred by its internal company name of M201 by Hemarina SA (Morlaix, France). HEMOXCell® (MOX) was extracted under gentle agitation of frozen Nereis virens at 4 °C followed by several purification steps and then conditioned in cryotubes. The final product was conserved at −80 °C and thawed at 4 °C before experimentation. HEMOXCell® catches the oxygen and releases it according to the partial pressure of oxygen (pO2) with an affinity (P50) such that half the binding sites would be oxygenated when under an oxygen pressure of about 37 mmHg at 37 °C (i.e. 5% O2) [Citation36]. The link with oxygen, on of oxyhemoglobin, is a cooperative and allosteric process affected by the saturation of oxygen and characterized by a cooperativity coefficient (n50) of about 1.47 at 37 °C [Citation36]. An optimal concentration of 0.025 g/L as previously found and described was used in our experiments [Citation38].
In this report, cell culture performed in control medium Dulbecco’s Modified Eagle’s medium (DMEM) and 0.025 g/L HEMOXCell®-supplemented medium were referred as DMEMCtl and DMEMMOX, respectively. Likewise the osteo-induction medium w/o and w/HEMOXCell® were referred to OBSCtl and OBSMOX.
Isolation and culture of hMSCs
MSCs were extracted from human bone marrow. Samples were obtained from patients who underwent total hip replacement surgery at the Department of Orthopedic Surgery at the Brest University Hospital (Brest, France). Before sampling, written consents of the patients were obtained and all precautions were taken to preserve the privacy of the donors and the project was approved by local ethical committees (ministerial authorization: DC-2009-1006).
Cells from bone marrow were isolated and characterized according to ISCT criteria as previously described [Citation38]. Cultures were performed into basic growth media consisting of alpha-minimum essential medium (α-MEM) (Lonza, Basel, Switzerland) containing 5% hPL, 1% l-glutamine 200 mM (Gibco®, France), 1% Ciprofloxacin 2 mg/mL (Panpharma®, France) and 0.04% Heparin (25,000 UI/5 ml) (Panpharma®, France) and maintained in a humidified atmosphere with 5% CO2 at 37 °C and the medium was changed twice a week thereafter. Adherent cells were cultured until passage two or three and aliquots were used to characterize them by their typical membrane expression markers and their trilineage differentiation ability before use [Citation38]. All experiments were conducted in triplicate with at least 3 independent assays.
Preparation of hPL
hPL was obtained from apheresis platelet concentrate pools (5 donors) provided by the French blood bank or "Etablissement Français du Sang" (Bretagne, Brest, France) after their expiration date and were biologically qualified according to the French legislation. The mean amount of platelets in each pool was >4 × 1011. hPL containing platelet-released growth factors was generated from platelet units through a repeated freezing–thawing procedure. Platelet bodies were removed by centrifugation (15 min, 1500 × g), and the supernatant was stored at −20 °C until use.
Analysis of cell osteogenic differentiation capacity
Osteogenic differentiation was induced in MSC cultures between passages 1 and 3. Cells reaching ∼80% confluence were trypsinized and plated in 24-well plates at a density of 5000 cells/well in duplicates and cultured for 24 h in the standard differentiation medium (DMEMCtl) consisting of DMEM high-glucose (4.5 g/L) (Dulbecco’s Modified Eagle’s Medium) (Lonza, Basel, Switzerland) supplemented the same as the already listed α-MEM medium. After this adherence period, the medium was replaced with the differentiation medium (OBSCtl) (i.e. standard medium supplemented with 1 µM dexamethasone, 0.05 mM ascorbic acid and 10 mM β-glycerophosphate (Sigma®, St Quentin-Fallavier, France)) for induced wells. The remaining wells were left without any additives and were cultured as an undifferentiated control. After 5, 10, 15 and 20 days of induction, alkaline phosphatase (ALP) activity and mineralization were analyzed. These experiments were performed with DMEM and OBS media w and w/o 0.025 g/L HEMOXCell® in duplicate for 3 different MSCs samples.
ALP activity: Osteoblastic phosphatase activity was visualized using the BCIP/NTB substrate (SigmaFastTM BCIP-NBT, Sigma®, France). Cells were fixed with 4% PFA for 10 min and rinsed with a washing buffer (0.05% Tween 20 in Phosphate Buffered Saline (PBS)). Fixed cell monolayers were incubated for 10 min with a substrate solution of 5-bromo-4-chloro-3-indolyl-phosphate-nitro blue tetrazolium) (BCIP-NBT) dissolved in distilled water. Enzymatic activity was visualized as a blue-violet stain.
Alizarin red staining: Extracellular calcium deposits were revealed using an orange-red stain with Alizarin Red S (Sigma®, France). Briefly, cells were fixed with 4% paraformaldehyde (PFA) for 15 min, followed by incubation in Alizarin Red S staining solution (20 mg/mL, pH 4.1–4.3) in the dark at room temperature (RT) for 30 min with gentle shaking. The staining solution was carefully aspirated and the cell monolayer was washed 3–4 times with distilled water and conserved in PBS.
Scaffolds design
Powder and cylinders of human cancellous bone (BioBank®, Presles-en-Brie, France) were used as scaffolds for cell proliferation in these experiments. The powder form was about 0.5 mm of diameter with a porosity of 80% and the bone cylinders (8 mm ×4 mm) had an approximate porosity of 70% and pore sizes ranging from 200 to 800 μm. These scaffolds were viro-inactivated and sterilized using the Supercript® process which is based on the use of supercritical CO2 (CO2SC). In this condition, CO2 behaves as a natural solvent for lipid removal.
Assessment of MSC compatibility with the BIOBank® substitute
To assess cell compatibility with bone scaffolds, MSCs were seeded on BIOBank® bone powder (1 × 106 cells/80 mg) on 15 ml Falcon® tubes in DMEM standard medium in static culture. Cultures were maintained during 23 days in standard condition (5% CO2, 37 °C) and were analyzed at 2, 10 and 23 days by confocal microscopy and SEM (scanning electron microscopy). For SEM, samples were incubated in 2.5% (w/v) glutaraldehyde solution for 1 h at room temperature (RT) and then washed three-times in PBS. Samples were dehydrated by a series of ethanol solutions. Specimens were observed under SEM (Hitachi S-3200 N).
Viability was assessed with Calcein AM and Ethidium homodimer (EthD-1) from the Live/Dead® Viability/Cytotoxicity Kit (MolecularProbes®, LifeTechnologiesTM) according to the user manual. Seeded powders were washed with PBS and incubated for 30 min at RT with 2 µM Calcein AM and 1.25 µM EthD-1 diluted in α-MEM medium. Constructs were observed with a fluorescence microscope (Axio Imager M2, Zeiss) (Calcein: 530 ± 12.5 nm; EthD-1: 645 ± 20 nm).
Analysis of cell proliferation on 3-D bone scaffolds
MSCs were seeded on bone scaffolds (5·106 cells/cm3) under static conditions, after 1 h incubation, constructs were maintained in a standard medium for 24 h and then media were replaced with DMEMCtl, DMEMMOX, OBSCtl or OBSMOX to a final volume of 500 µl. Cell number was calculated using Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Rockville, MD) according to the manufacturer’s instructions. Briefly, after 7 and 14 days of culture, 50 µl of CCK-8 solution was added to constructs medium and incubated for 2 h at 37 °C. Then 100 µl from each condition were dispensed into a 96-well plate. The optical density was then measured at 450 nm using a microplate reader. Cell quantity was determined using a standard curve. Unseeded scaffolds were used as controls.
Static and perfusion culture on bone cylinders
Experimental design
MSCs were cultured with bone cylinders using two different cultivation methods. A static culture was performed in 6-well plates. Scaffolds were seeded by the injection of 1 × 106 cells in DMEM standard medium and then incubated in standard condition for 1 h to allow cell attachment. Scaffolds were then cultured in DMEMCtl or DMEMMOX. Perfused scaffolds were seeded with 1 × 106 cells using a perfusion bioreactor system (U-Cup®, CellecBiotek, Basel, Switzerland) described in previous work [Citation39]. According to the user’s manual, a seeding period of 24 h with a flow rate 3 ml/min was used to calculate the seeding efficiency. During the culture, perfusion rate was reduced to 0.3 ml/min.
For the cultivation period, four different media were used: DMEMCtl, DMEMMOX, OBSCtl and OBSMOX. The media were changed twice a week and an aliquot from each medium change was collected and stored at −20 °C for ALP dosage. For experiments, constructs were cultured either statically or perfused for a 4 week period.
Cell seeding efficiency
Cell seeding efficiency in the scaffolds was investigated for both static and perfusion seeding methods after 24 h of culture. Medium was removed from the wells and perfusion systems. For the static conditions, adherent cells on the bottom wells were trypsinized and collected with cells remaining in medium. Total cell number was evaluated using an ADAM automated cell counter (Labtech, France). Seeding efficiency was calculated as a percentage of seeding cells among the total cell number (i.e. 1·106 cells/scaffolds). This experiment was performed in triplicate (n = 3).
Histological analysis
Scaffolds were fixed for 24 h at 4 °C and then dehydrated in a graded series of ethanol baths for four days. After dehydration, the bone scaffolds were infiltrated and embedded in methyl methacrylate (MMA) resin for 1 week (Technovit 9100 NEW, Kulzer; Hanau, Germany). Then, serial 5 mm deep sections were cut from the cylinders along its perpendicular axis using a diamond saw made for undecalcified tissues (Polycut Leica SM2500, Leica Biosystems; Wetzlar, Germany). Resin was removed from the bone sections using acetone and sections were stained with Movat’s pentachrome and Hematoxylin-Eosine-Safran (HES) using the Varistain™ Gemini ES Automated Slide Stainer (Thermo ScientificTM) and observed under a light microscope (Axioplan 2, Zeiss; Jena, Germany).
Immunostaining of Collagen I (Col I) was performed with an anti-human Col I antibody (ab138492, 1/250, Abcam, Cambridge, UK) on sections after incubation with acetone to remove embedding resin and with 3% peroxide hydrogen (Sigma®) to block endogenous peroxidases. Incubation with biotinylated secondary antibody (anti-rabbit, Dako) was performed and Col I positive staining was developed in brown colour using the LSAB®2 System-HRP Kit (Dako). Mayer’s Hematoxylin (Sigma®) was used as a counterstain. Slides were mounted in Eukitt® medium and observed under a light microscope (Axioplan 2, Zeiss; Jena, Germany).
SEM analysis
Scaffolds cultured in all conditions were treated for SEM analysis. In the same way as described in the section titled “Cell morphology and viability assays on scaffold powder”. Briefly, samples were incubated in 2.5% (w/v) glutaraldehyde solution for 2 h at room temperature (RT) and washed three-times in PBS. Partial decalcification was performed using Super Decalcifier I solution (Polysciences, Le Parray-en-Yvelines France) for 30 min at RT. Decalcified cylinders were rinsed three times with PBS and then cut into two parts vertically to analyze inner and outer cellularization. Samples were then dehydrated by a series of ethanol solutions and observed under SEM (Hitachi S-3200 N).
MSC RNA isolation and qRT-PCR
Four osteogenic markers were evaluated through quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) at day 14 and day 21 of culture, in order to analyze the impact of 0.025 g/L MOX on MSC osteoblastic differentiation under static and perfused BIOBank® scaffolds. Total RNA was isolated from the MSCs using the RNeasy Mini Kit (Qiagen, Courtaboeuf, France). RNA was then reverse transcribed to complementary DNA (cDNA) using a SuperScript™ II Reverse Transcriptase Kit (Life Technologies) in accordance with the manufacturer’s protocol; 500 ng of cDNA were amplified using QuantiTect® SYBR® Green PCR Kit (QIAGEN®, Courtaboeuf, France) with LightCycler® 480 Instrument II (Roche, Meylan, France). Primers were provided by Eurogentec (Angers, France) (NCBI reference number in brackets): alpha-1 type I collagen (COL1A1) [NM_000088], ALP [NM_000478.4], osteopontin (OPN) [NM_001251830], RUNX2 [NM_001024630] and the endogenous gene control ribosomal protein L13a (RPL13A) [NM_001270491]. Ct average was determined from raw data and the relative gene expression level of each target gene was normalized to the mean of RPL13A in each group. The fold change was then determined relative to MSCs control cultured in 2-D DMEM medium. Relative quantification was performed using the ΔΔCt method. Samples were completed in technical triplicates and mean ± standard deviations were reported. Unpaired T-test was used to highlight significant differences in gene expression (p values < .05).
Results
Effect of HEMOXCell® on MSCs osteogenic differenciation
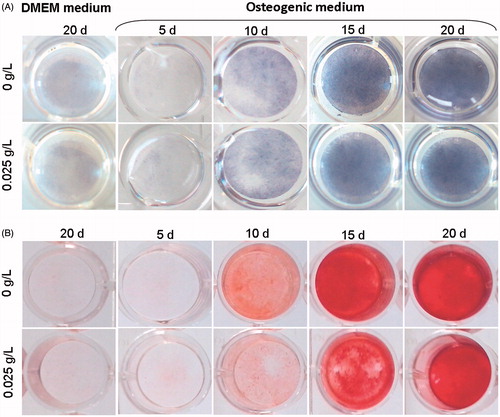
Osteoblast differentiation of MSCs was analyzed after 5, 10, 15 and 20 days of culture with ALP activity quantification and Alizarin red staining (). After 5 days of OBS medium treatment, no staining was observed for both experiments, indicating that cells were still not differentiated enough to express these markers. From the 10th day to the 20th day, a similar profile was obtained for both analyzes. The colour intensity of both OBSCtl and OBSMOX media increased in a time-dependent manner, corresponding to an increase of the ALP activity, simultaneously with the mineralized matrix production. The adjunction of HEMOXCell® to MSC osseodifferentiation medium does not seem to have any effect on bone marrow MSCs differentiation in PL-supplemented medium.
Figure 1. Kinetic analysis of MSC osteoblast differentiation with HEMOXCell®. Osteoblast differentiation was analyzed at 5, 10, 15 and 20 days of culture in OBSCtl and OBSMOX media. DMEMCtl and DMEMMOX were used as undifferentiated references. Images represented (A) ALP activity staining of osteoblasts and (B) Alizarin red staining of mineralized matrix. Representative results of 3 independent experiments are shown.
Analysis of MSC/scaffold compatibility
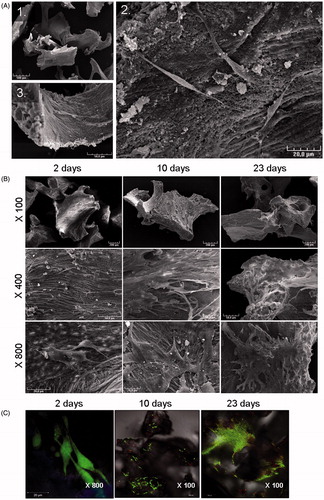
The SEM analysis of seeded bone powder showed an irregular structure with an average diameter of 500 µm and adherent MSCs exhibited a fibroblast-like morphology (). After the first 24 h, MSCs already showed adhesion spreading processes demonstrating the cytocompatibility of the scaffold. The adherence of MSCs on BIOBank® substitute for a longer culture period, under static condition, was then investigated as well as their viability ()). SEM analysis after 2, 10 and 23 days showed a strong increase in adherent cells number during the culture period (). After 23 days, cells were confluent and covered the entire surface of the scaffold. The Live/Dead® staining revealed a high cell viability (green cell staining) until day 23 (). However, no evidence of cell death (red cell staining) was observed in any condition. These results suggest the ability of MSCs to adhere and proliferate on the BIOBank® substitute, and show the non-cytotoxicity of the scaffold.
Figure 2. Analysis of cell viability and proliferation. (A) Construct morphology was observed using scanning electron microscopy. SEM microphotographs of BIOBank® powder cultured 24 h with MSCs. Scale bars represent 500 µm (1), 20 µm (2) and 50 µm (3). MSCs adhere to the scaffold and exhibit a fibroblast-like morphology. (B,C) Morphology and viability were analyzed after 2, 10 and 23 days. (B) Representative SEM images of MSCs cultured on bone powder at different magnifications, from top to bottom: ×100, ×400, ×800. (C) Confocal microscopy analysis of MSCs viability using Live/Dead® staining showed cell proliferation and viability on the powder. Data are representative of 3 different experiments.
Cell proliferation
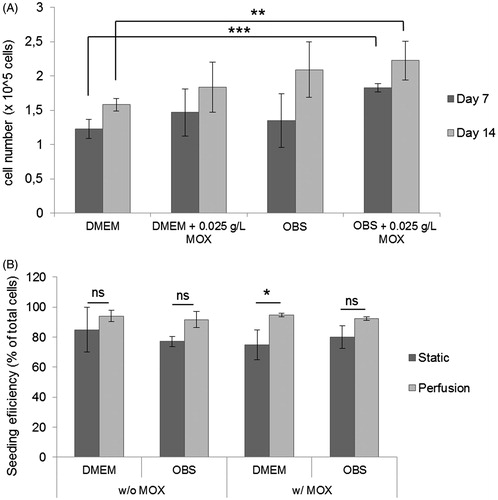
A static analysis of cell proliferation was performed with the CCK-8 proliferation assay, in order to analyze the cellular proliferation potential on the cylinder scaffolds with the oxygen carrier. In all tested conditions, we obtained an increase in cell density between 7 and 14 days of culture (). A significantly higher cell density was obtained with OBSMOX when compared to DMEMCtl after 7 days (p < .001) and after 14 days (p = .0087), indicating that MSC proliferation in 3-D scaffolds under static condition was promoted in differentiation media supplemented with the oxygen carrier, HEMOXCell®.
Figure 3. Cell proliferation and scaffolds seeding efficiency analysis. (A) CCK-8 proliferation assay of cell-constructs was performed under different conditions after 7 and 14 days under static culture. Data represent mean ± SD (n = 4, **p < .01; ***p < .0001; ns: non-significant). (B) MSCs were seeded into bone cylinders and cultured under static and perfusion in DMEM and osteogenic media (with and without MOX). After 24 h, seeding efficiency was calculated. For each condition, 10 different scaffolds were analyzed. Data are represented as mean ± SD (**p < .01).
Comparison of static and perfusion culture with HEMOXCell®
Analysis of the seeding efficiency
At the beginning of the culture period, the seeding efficiency was investigated for both static and hydrodynamic culture systems in all culture media (). The static seeding technique was performed by deposition of 1 × 106 cells per cylinder and resulted in 84.97 ± 14.95% and 74.78 ± 9.87% cell seeding efficiency for DMEMCtl and DMEMMOX, respectively and 77.2 ± 3.89% and 80.01 ± 7.27% for OBSCtl and OBSMOX. Better results were obtained for the perfusion seeding method with a high reproducibility between DMEM and OBS conditions (DMEMCtl and DMEMMOX: 94.07 ± 3.75% and 94.87 ± 1.18%, respectively; OBSCtl and OBSMOX: 91.72 ± 5.43% and 92.42 ± 1.27%). These results suggested that the perfusion seeding method equally promote the cellular adherence w/and w/o 0.025 g/L MOX.
Histological analysis
Movat and HES staining were performed on undecalcified bone sections to analyze the preservation of the cellular structures when using a resin embedding method for histological analysis. ) showed representative micrographs of Movat and HES staining. Only a few cells were observed under static culture conditions whereas some large cell clusters were observed in perfused scaffolds in OBS medium. The creation of ECM was observed in high magnitude for OBSCtl and OBSMOX perfused scaffolds (orange/pink and blue colours for HES and Movat staining, respectively). Images of entire sections clearly showed the scaffolds filled with cells when using OBS media. These cells were more numerous compared to other test conditions.
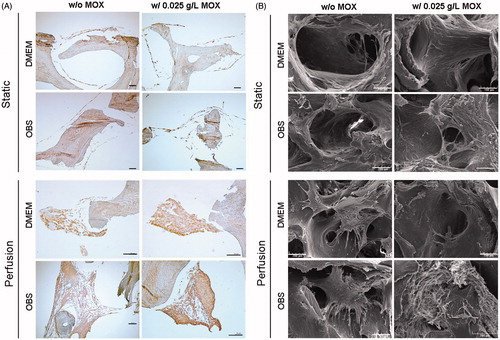
Figure 4. Histological analysis of cell distribution throughout BIOBank® scaffolds on resin embedding sections. Representative histological images of undecalcified scaffolds for cultures performed under static or perfusion systems after 4 weeks and stained with HES (A) and Movat’s Pentachrome (B) (Specimens diameter represents 8 mm). (HES stains: blue/black = nuclei; orange/pink = collagen; pink = cytoplasm) (Movat stains: red = fibrin; blue = mucin and extrafibrillar matrix; black = nuclei; yellow = collagen).
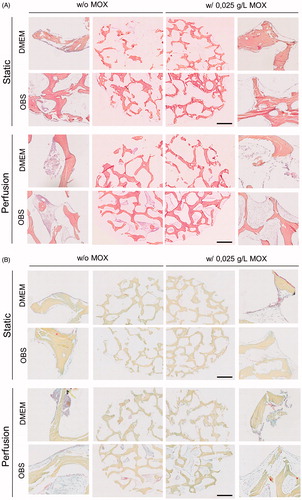
On both culture systems, MSCs adhered to the scaffold surfaces, but perfusion culture allowed the formation of multiple cell layers. Sectioning performed with this method lead a detachment of the cell layers from the bone. According to the results obtained, cell proliferation within the scaffold pores under perfusion culture appeared to be increased and more homogeneous when using HEMOXCell® as supplement.
The presence of Col I was investigated by immunohistochemistry and resulted in a brown uniform colour. Col I labelling was detected in all conditions (). A stronger expression was observed in perfusion cultures indicating the effect of the flow perfusion on the ECM production. A brown colouration of the bone scaffolds was also observed due to collagenic composition of the bone matrix.
Figure 5. Immunohistological staining of Col I and scanning electron micrographs. (A) Representative images of Col I immunostaining for cells cultures under static and perfusion with and without 0.025 g/L HEMOXCell®. Data showed a high expression of Col I (brown stain) by MSCs especially under perfusion culture. Scale bars represent 100 µm. (B) SEM analysis of cell-scaffold constructs was performed after in vitro culture for 4 weeks. Images represent a magnification on a pore (scale bars represent 100 µm). Images are representative of two experiments.
Scanning electron microscopy analysis
Morphologies of the cellular structures within the scaffold pores were examined after 4 weeks of culture using SEM (). Under static culture, the results obtained from DMEMCtl and DMEMMOX showed a cellular monolayer lining the scaffold pores and a thick cell layer on the scaffold surfaces. For OBSCtl and OBSMOX, the cell growth was more extensive and formed multilayer structures. In comparison with static culture condition, cellular density appeared significantly greater for the perfused group. Cells exhibited large filopodia structures forming an interaction network between their membranes and scaffold. Large 3-D cellular structures were observed in all conditions, which filled a part of the scaffold pores for OBSCtl and OBSMOX. Perfusion culture seems to promoted cell density as well as cell–cell interactions. A lower scaffold cellularization was obtained for DMEM conditions in both static and perfusion systems. The use of HEMOXCell® seems to improve multilayer structure formation especially under perfusion of OBS medium. Furthermore, a crumbling effect was observed in all constructs processed with the undecalcifying method.
MSC RNA isolation and qRT-PCR
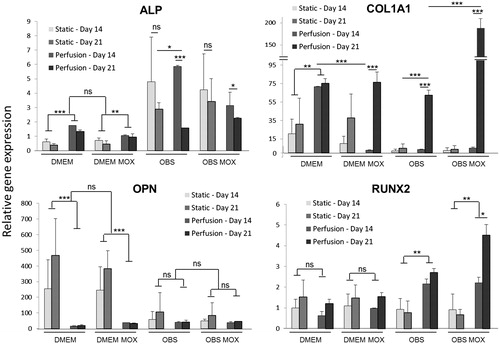
Gene expression analysis of osteogenic markers was conducted on days 14 and 21 after static and perfusion cultures on DMEM and OBS media (w/and w/o MOX) (). In this study ALP, COL1A1, OPN and RUNX2 gene expression values were specifically normalized to MSCs cultured in 2-D in DMEM. ALP gene expression was significantly lower for static DMEM cultures for both 14 and 21 days compared with perfusion DMEM groups. In OBS media, mRNA levels were up-regulated in comparison with DMEM media. No differences were observed between 14 and 21 days of culture under OBS static conditions. However, ALP levels significantly decreased at day 21 in OBS perfused cultures compared to day 14. No impact of MOX supplementation was detected. In the same way, no significant differences of Col1A1 mRNA levels were observed after 14 and 21 days of culture under static conditions. However, values were higher in DMEM media compared with OBS media. Under perfusion Col1A1 mRNA was significantly up-regulated after 21 days of culture (p < .001) for DMEMMOX, OBSCtl and OBSMOX. The highest value was obtained for perfused OBSMOX group at day 21. Conversely, significant elevated OPN mRNA levels (p < .001) were observed in cells that were statically cultured in DMEM medium (w/and w/o MOX) for both 14 and 21 days compared to the perfused cultures in all conditions. Interestingly, no major change in mRNA levels was observed between perfused samples as well as between cultures performed in OBS media. The expression of RUNX2 remained low throughout the study excepted for OBS perfusion groups which display a significant up-regulation on day 21 compared to day 14 especially for OBSMOX group. Similar levels were maintained for all the other groups.
Figure 6. Evaluation of MSCs osteoblastic differentiation in bone cylinders. Osteogenic markers (ALP, COL1A1, OPN, RUNX2) expression levels were quantified at day 14 and 21 for MSCs grown in 3D-scaffolds with DMEM and OBS media (with and without MOX) under static and perfused conditions. qPCR data are represented as fold-change compared with the expression levels found in the uninduced MSCs cultured in 2-D. Values are mean ± SD (n = 3). Highly significant differences between 14 days and 21 days and between condition groups are represented with asterisks (*p < .05, **p < .001 and ***p < .0001).
Discussion
Bone regeneration is clinically relevant especially in dental surgery and represents a great challenge for surgeons and researchers [Citation1,Citation3,Citation40]. Tissue engineering of bone aims to create a viable substitute in vitro with the combination of osteocompetent cells with a biocompatible scaffold in order to facilitate new bone formation into the injured tissue [Citation19].
Various factors are involved in the success of bone tissue engineering applications, including the scaffold, the cell inoculum, the cell culture medium, soluble molecules as well as the cell spatial distribution [Citation28]. Static culture is the most commonly used strategy for cell culture in planar and 3-D cultures. However, static culture has been largely documented to be not effective at distributing cells through the scaffold [Citation31,Citation41–43]. Indeed, gradients of oxygen and nutrients often take place within the constructs, resulting in spatial variations in cell density and viability [Citation44]. In the present study, we compared the effects of static and perfusion methods in the seeding efficiency and proliferation of MSCs into 3-D porous bone substitutes, using MOX as oxygen carrier in LP-supplemented medium.
MSCs were selected for these experiments because of their numerous properties that make them the most promising candidates for bone tissue engineering [Citation19]. In this work, we firstly investigated the osteoblastic differentiation of MSCs using MOX in a time-dependent manner (). Similar differentiation profiles were obtained for ALP activity and mineralized matrix production, w/and w/o MOX, supporting the potential use of this supplement for bone tissue engineering. We then investigated the use of a porous human bone substitute, to support MSCs 3-D culture under standard (DMEM) and osteoblastic (OBS) environment.
The compatibility of the scaffold and its ability to support cellular proliferation with DMEMCtl and DMEMMOX media were observed by SEM and confocal analysis of MSCs seeded on a powder form of the bone scaffold (). Cell proliferation measurements supported the feasibility of associating MSCs with the Biobank® substitute in the presence of MOX in an osteogenic environment (). Concerning the bone cylinders used on this work, we found no significant differences between the efficiencies of the two seeding methods tested (i.e. static and perfusion) (). It was reported that the initial cell seeding density of MSCs has an effect on bone tissue formation in vitro [Citation11]. In our study, we used an initial cell seeding density of 1 million cells/100 mg scaffold, which is in accordance with other studies showing that a higher cell density has no beneficial effects on the bone forming abilities [Citation11,Citation45].
In this work, constructs were maintained for 4 weeks in culture and analyzed using histological techniques on resin embedded sections. A cellular structure detachment was observed as well as a crumbing of the bone structure due to the natural stiffness of the bone tissue.
The perfusion flow rate in bioreactors directly impacts the oxygen supply in depth [Citation46]. However, the flow rate also has an effect on differentiation as well as on cell detachment. In this study, we worked at a slow perfusion rate (0.3 ml/min) to avoid a large shear stress while still prevent a nutrition gradient formation. This low perfusion rate was found to be optimal for the scaffolds’ cellularization. In addition, we compared different culture conditions to analyze cell behaviour under an osteogenic environment and with the contribution of HEMOXCell® in the 3-D culture of MSC. A 3-D culture under perfusion system was demonstrated to be the more effective than static system for cell proliferation among all the tested conditions, with a higher density and a more uniform spatial distribution of the cells. These points have been largely discussed on literature as well as its implication on MSC osteogenic differentiation [Citation31,Citation33,Citation47,Citation48]. In conventional cell culture method, the cell distribution is limited to the periphery of the scaffold with few cells observed in the inner part of the scaffolds.
Scaffolds cultured statically in DMEM medium (w/or w/o MOX) remained equally cellularized with adherent cell layers. Conversely, perfusion strongly promoted scaffold cellularization especially under osteogenic condition. Additionally, histological and SEM results revealed a positive impact of MOX supplementation in this condition. It is known that differentiation medium highly stimulates cell proliferation as it was the case in our analyses [Citation49]. The beneficial effect of MOX supplementation under osteogenic condition could be explained by the increase of cell metabolism during differentiation inducing a higher oxygen consumption [Citation50]. In this context, HEMOXCell® acts by releasing oxygen according the gradient created by cell consumption through the medium to restore oxygen basal level. Furthermore, it has been shown that pO2 level in pericellular environment can be hypoxic in case of a high cell density and high metabolic rates [Citation51].
Concerning the effect of HEMOXCell® on the differentiation of hMSCs cultured on 3-D scaffolds, the expression of osteogenic biomarkers ALP, COL1A1, OPN and RUNX2 was investigated under static and perfusion cultures (). qPCR results demonstrate that MOX supplementation does not appear to strongly impact the osteogenesis of MSCs in vitro. However, after 21 days of perfusion culture in OBS medium significant higher COL1A1 and RUNX2 transcript levels were observed. Col I is a natural component of stromal cells, ECM and bone matrix. In all samples, MSCs secreted a matrix containing Col I with higher colour intensity for perfused scaffolds with HEMOXCell® () supporting qPCR data (). RUNX2, which was described to be important for the osteogenic differentiation [Citation52], was also up-regulated under perfusion culture in OBS medium according to culture time especially with MOX. Additionally, ALP was analyzed as a biomarker of mature osteogenesis and was observed to peak in expression level faster when cells were cultured under differentiation media as previously described [Citation53]. In any event, post-translational modification of osteogenic markers is a critical step in the differentiation of MSCs and had to be further considered [Citation54]. In addition, it was previously reported that the use of hPL promotes MSCs osseodifferentiation [Citation55,Citation56]. As limitations, only four osteogenic markers were investigated in our study and their association on MSC bone forming capacity in vivo is still unclear.
The results obtained showed the beneficial role of perfusion culture in ECM production as well as for MSCs osteoblastic differentiation in HEMOXCell®-supplemented media. These results clearly suggest that the use of HEMOXCell® as an additive to the differentiation culture media does not negatively impact the differentiation process.
In this present study, we evaluated the impact of this marine oxygen carrier in the culture of human MSCs on BIOBank® bone substitute for bone tissue engineering applications. This haemoglobin is a GMP-compliant product usable for medical purposes with a high oxygen carrying potential and a SOD-like activity [Citation35]. Sensitivity of HEMOXCell® to oxygen variations constitute an interesting tool for bone tissue engineering by promoting scaffold cellularization in static conditions and even under perfusion in vitro. HEMOXCell® could potentially be used to promote the oxygenation of tissue substitutes in depth after implantation, and therefore the tissue repair.
| Abbreviations | ||
| 3-D | = | three-dimensional |
| ALP | = | alkaline phosphatase |
| BCIP | = | 5-bromo-4-chloro-3-indolyl-phosphate |
| CHO | = | Chinese hamster ovary |
| CO2 | = | carbon dioxide |
| Col I | = | collagen I |
| DFDBA | = | demineralized freeze-dried bone allografts |
| DMEM | = | Dulbecco modified Eagle’s minimal essential medium |
| ECM | = | extracellular matrix |
| EGF | = | epidermal growth factor |
| EthD-1 | = | ethidium homodimer |
| FBS | = | foetal bovine sera |
| FDBA | = | freeze-dried bone allograft |
| hPL | = | human platelet lysate |
| H&E | = | hematoxylin and eosin |
| HES | = | hematoxylin-eosin-safran |
| HRP | = | horseradish peroxidase |
| LSAB | = | labelled streptavidin biotin |
| α-MEM | = | minimum essential medium alpha modifications |
| MMA | = | methyl methacrylate |
| MOX | = | HEMOXCell |
| MSC | = | mesenchymal stem cell |
| O2 | = | molecular oxygen |
| OBS | = | osteoblastic |
| PBS | = | phosphate-buffered saline; |
| PDGF | = | platelet-derived growth factor |
| PO2 | = | partial pressure of oxygen |
| NBT | = | nitro blue tetrazolium |
| PFA | = | paraformaldehyde |
| RT | = | room temperature |
| SEM | = | scanning electron microscopy |
| SOD | = | superoxide dismutase |
| TGF | = | transforming growth factor beta 3 |
| VEGF | = | vascular endothelial growth factor |
Acknowledgements
We are grateful to Raphaël Bardonnet (Chief Scientific Officer at BIOBank®, France) for his material support and his implication in this project. We are grateful to Professor Jérôme Guicheux (LIOAD, Nantes) for his scientific contribution. Special thanks to Gérard Sinquin and Philippe Elies (Plateforme d'Imagerie et de Mesures en Microscopie, PIMM, Faculty of Sciences, Brest) for SEM and confocal analysis.
Disclosure statement
FZ is founder of and hold stock in HEMARINA SA, which produces the substance being investigated. EL holds stocks in HEMARINA SA. FL was an employee of HEMARINA SA and does not hold stock. All other authors declare no conflict of interest. HEMOXCell® samples were provided by HEMARINA SA.
Additional information
Funding
References
- Xu B, Zhang J, Brewer E, et al. Osterix enhances BMSC-associated osseointegration of implants. J Dent Res. 2009;88:1003–1007.
- Fuerst G, Gruber R, Tangl S, et al. Enhanced bone-to-implant contact by platelet-released growth factors in mandibular cortical bone: a histomorphometric study in minipigs. Int J Oral Maxillofac Implants. 2003;18:685–690.
- Vandamme K, Holy X, Bensidhoum M, et al. In vivo molecular evidence of delayed titanium implant osseointegration in compromised bone. Biomaterials. 2011;32:3547–3554.
- Ihde S, Kopp S, Gundlach K, et al. Effects of radiation therapy on craniofacial and dental implants: a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:56–65.
- Vandamme K, Holy X, Bensidhoum M, et al. Establishment of an in vivo model for molecular assessment of titanium implant osseointegration in compromised bone. Tissue Eng Part C Methods. 2011;17:311–318.
- Giannoudis PV, Dinopoulos H, Tsiridis E. Bone substitutes: an update. Injury. 2005;36(Suppl.3):S20–S27.
- Bostrom MPG, Seigerman DA. The clinical use of allografts, demineralized bone matrices, synthetic bone graft substitutes and osteoinductive growth factors: a survey study. HSS J. Musculoskelet. J Hosp Spec Surg. 2005;1:9–18.
- Fretwurst T, Spanou A, Nelson K, et al. Comparison of four different allogeneic bone grafts for alveolar ridge reconstruction: a preliminary histologic and biochemical analysis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:424–431.
- Mitton D, Rappeneau J, Bardonnet R. Effect of a supercritical CO2 based treatment on mechanical properties of human cancellous bone. Eur J Orthop Surg Traumatol. 2005;15:264–269.
- Scherberich A, Galli R, Jaquiery C, et al. Three-dimensional perfusion culture of human adipose tissue-derived endothelial and osteoblastic progenitors generates osteogenic constructs with intrinsic vascularization capacity. Stem Cells Dayt Ohio. 2007;25:1823–1829.
- Holy CE, Shoichet MS, Davies JE. Engineering three-dimensional bone tissue in vitro using biodegradable scaffolds: investigating initial cell-seeding density and culture period. J Biomed Mater Res. 2000;51:376–382.
- Daar AS, Greenwood HL. A proposed definition of regenerative medicine. J Tissue Eng Regen Med. 2007;1:179–184.
- Friedenstein AJ, Chailakhyan RK, Latsinik NV, et al. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. 1974;17:331–340.
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317.
- Arthur A, Zannettino A, Gronthos S. The therapeutic applications of multipotential mesenchymal/stromal stem cells in skeletal tissue repair. J Cell Physiol. 2009;218:237–245.
- Segawa Y, Muneta T, Makino H, et al. Mesenchymal stem cells derived from synovium, meniscus, anterior cruciate ligament, and articular chondrocytes share similar gene expression profiles. J Orthop Res. 2009;27:435–441.
- Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36:568–584.
- Arvidson K, Abdallah BM, Applegate LA, et al. Bone regeneration and stem cells. J Cell Mol Med. 2011;15:718–746.
- Grayson WL, Bunnell BA, Martin E, et al. Stromal cells and stem cells in clinical bone regeneration. Nat Rev Endocrinol. 2015;11:140–150.
- Wang X, Wang Y, Gou W, et al. Role of mesenchymal stem cells in bone regeneration and fracture repair: a review. Int Orthop. 2013;37:2491–2498.
- Fu W-L, Xiang Z, Huang F-G, et al. Coculture of peripheral blood-derived mesenchymal stem cells and endothelial progenitor cells on strontium-doped calcium polyphosphate scaffolds to generate vascularized engineered bone. Tissue Eng. Part A. 2015;21:948–959.
- Li H, Dai K, Tang T, et al. Bone regeneration by implantation of adipose-derived stromal cells expressing BMP-2. Biochem Biophys Res Commun. 2007;356:836–842.
- Yoon E, Dhar S, Chun DE, et al. In vivo osteogenic potential of human adipose-derived stem cells/poly lactide-co-glycolic acid constructs for bone regeneration in a rat critical-sized calvarial defect model. Tissue Eng. 2007;13:619–627.
- Chen X, Armstrong MA, Li G. Mesenchymal stem cells in immunoregulation. Immunol Cell Biol. 2006;84:413–421.
- Chevallier N, Anagnostou F, Zilber S, et al. Osteoblastic differentiation of human mesenchymal stem cells with platelet lysate. Biomaterials. 2010;31:270–278.
- Wang D, Jiang H, Wang S, et al. Construction of tissue-engineered bone using a bioreactor and platelet-rich plasma. Exp Ther Med. 2014;8:413–418.
- Trouillas M, Prat M, Doucet C, Ernou I, Laplace-Builhé C, Blancard PS, etet al. A new platelet cryoprecipitate glue promoting bone formation after ectopic mesenchymal stromal cell-loaded biomaterial implantation in nude mice. Stem Cell Res Ther. 2013;4:1.
- Lovett M, Lee K, Edwards A, et al. Vascularization strategies for tissue engineering. Tissue Eng Part B Rev. 2009;15:353–370.
- Leotot J, Coquelin L, Bodivit G, et al. Platelet lysate coating on scaffolds directly and indirectly enhances cell migration, improving bone and blood vessel formation. Acta Biomater. 2013;9:6630–6640.
- Ng F, Boucher S, Koh S, et al. PDGF, TGF-β, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood. 2008;112:295–307.
- Kasper FK, Liao J, Kretlow JD, et al. Flow perfusion culture of mesenchymal stem cells for bone tissue engineering. StemBook [Internet]. Cambridge (MA): Harvard Stem Cell Institute; 2008 [cited 2015 Sep 8]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK27082/
- Bancroft GN, Sikavitsas VI, van den Dolder J, et al. Fluid flow increases mineralized matrix deposition in 3D perfusion culture of marrow stromal osteoblasts in a dose-dependent manner. Proc Natl Acad Sci USA. 2002;99:12600–12605.
- Papadimitropoulos A, Piccinini E, Brachat S, et al. Expansion of human mesenchymal stromal cells from fresh bone marrow in a 3D scaffold-based system under direct perfusion. PLoS One. 2014;9:e102359.
- Chabasse C, Bailly X, Rousselot M, et al. The multigenic family of the extracellular hemoglobin from the annelid polychaete Arenicola marina. Comp Biochem Physiol B, Biochem Mol Biol. 2006;144:319–325.
- Rousselot M, Delpy E, Drieu La Rochelle C, et al. Arenicola marina extracellular hemoglobin: a new promising blood substitute. Biotechnol J. 2006;1:333–345.
- Rousselot M, Dutheil D, Zal F. Novel heamoglobin and uses thereof [Internet]. 2010 [cited 2016 Jul 7]. Available from: http://www.google.com/patents/WO2010128159A1.
- Le Pape F, Bossard M, Dutheil D, et al. Advancement in recombinant protein production using a marine oxygen carrier to enhance oxygen transfer in a CHO-S cell line. Artif Cells Nanomed Biotechnol. 2015;43:186–195.
- Le Pape F, Cosnuau-Kemmat L, Richard G, et al. HEMOXCell, a new oxygen carrier usable as an additive for mesenchymal stem cell culture in platelet lysate-supplemented media. Artif Organs. 2017;41:359–371.
- Wendt D, Marsano A, Jakob M, et al. Oscillating perfusion of cell suspensions through three-dimensional scaffolds enhances cell seeding efficiency and uniformity. Biotechnol Bioeng. 2003;84:205–214.
- Nazirkar G, Singh S, Dole V, et al. Effortless effort in bone regeneration: a review. J Int Oral Health. 2014;6:120–124.
- Volkmer E, Drosse I, Otto S, et al. Hypoxia in static and dynamic 3D culture systems for tissue engineering of bone. Tissue Eng Part A. 2008;14:1331–1340.
- Pazzano D, Mercier KA, Moran JM, et al. Comparison of chondrogensis in static and perfused bioreactor culture. Biotechnol Prog. 2000;16:893–896.
- Liu C, Abedian R, Meister R, et al. Influence of perfusion and compression on the proliferation and differentiation of bone mesenchymal stromal cells seeded on polyurethane scaffolds. Biomaterials. 2012;33:1052–1064.
- Papadimitropoulos A, Scotti C, Bourgine P, et al. Engineered decellularized matrices to instruct bone regeneration processes. Bone. 2015;70:66–72.
- Luo F, Hou T-Y, Zhang Z-H, et al. Effects of initial cell density and hydrodynamic culture on osteogenic activity of tissue-engineered bone grafts. PLoS One. 2013;8:e53697.
- Volkmer E, Otto S, Polzer H, et al. Overcoming hypoxia in 3D culture systems for tissue engineering of bone in vitro using an automated, oxygen-triggered feedback loop. J Mater Sci Mater Med. 2012;23:2793–2801.
- Martin I, Wendt D, Heberer M. The role of bioreactors in tissue engineering. Trends Biotechnol. 2004;22:80–86.
- Gonçalves F, da C, Paz AH, et al. Dynamic culture improves MSC adhesion on freeze-dried bone as a scaffold for bone engineering. World J Stem Cells. 2012;4:9–16.
- Nishimura I, Hisanaga R, Sato T, et al. Effect of osteogenic differentiation medium on proliferation and differentiation of human mesenchymal stem cells in three-dimensional culture with radial flow bioreactor. Regen Ther. 2015;2:24–31.
- Pattappa G, Heywood HK, de Bruijn JD, et al. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J Cell Physiol. 2011;226:2562–2570.
- Metzen E, Wolff M, Fandrey J, et al. Pericellular PO2 and O2 consumption in monolayer cell cultures. Respir Physiol. 1995;100:101–106.
- Bruderer M, Richards RG, Alini M, et al. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. 2014;28:269–286.
- Prins H-J, Braat AK, Gawlitta D, et al. In vitro induction of alkaline phosphatase levels predicts in vivo bone forming capacity of human bone marrow stromal cells. Stem Cell Res. 2014;12:428–440.
- Ding H, Chen S, Yin J-H, et al. Continuous hypoxia regulates the osteogenic potential of mesenchymal stem cells in a time-dependent manner. Mol Med Rep. 2014;10:2184–2190.
- Arpornmaeklong P, Kochel M, Depprich R, et al. Influence of platelet-rich plasma (PRP) on osteogenic differentiation of rat bone marrow stromal cells. An in vitro study. Int J Oral Maxillofac Surg. 2004;33:60–70.
- López-Pérez PM, da Silva RMP, Sousa RA, et al. Plasma-induced polymerization as a tool for surface functionalization of polymer scaffolds for bone tissue engineering: an in vitro study. Acta Biomater. 2010;6:3704–3712.