
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of the present study was to prepare a novel CD133 aptamer modified DTX liposome system and investigate its characteristics in vitro and in vivo studies. In this study, the CD133-DTX LP was prepared by the thin-film hydration method and with the particle size of 100–120 nm. The TEM photomicrographs were smooth, sub-spherical in shape and aggregated to form small clusters. In vitro, a relatively slower DTX release profile was observed in CD133-DTX LP due to the presence of CD133 aptamers on the outer surface which might hinder the drug release. The drug release mechanism fit well with the Higuchi equation better. In cytotoxicity study, CD133 aptamers modified DTX LP significantly decreased cell proliferation and improved the therapeutic efficiency. In vivo imaging result indicated that CD133-DTX LP had very good tumour targeting ability. In vivo antitumour activity indicated that the CD133-DTX LP showed a significant antitumour activity in A549 tumour mice, with a very low systemic toxicity.
Introduction
Lung cancer is one of the most dangerous diseases in humans, accounts for nearly 15% of all new cancers, and causes more than one million deaths annually [Citation1,Citation2]. In 2016, lung cancer was the leading cause of cancer deaths in 65+ years in China [Citation3]. It is estimated that approximately 80% of lung cancer patients have a histologic diagnosis of non-small cell lung cancer (NSCLC) [Citation4]. Currently, the lung cancer survival is usually unsatisfactory by its recurrence, multi-drug resistance and metastasis [Citation5,Citation6]. As we all know, chemotherapy remains the primary treatment strategy for patients with NSCLC. However, the treatment effect of chemotherapy is limited due to serious systemic toxicity and limitations of the treatment range [Citation7]. Therefore, it is urgent to propose effective treatment for lung cancer.
Lung cancer stem cells (CSCs) are considered to initiate lung cancer [Citation8]. Some researchers hold the opinion that lung CSCs contribute to the recurrence and metastasis of lung cancer [Citation9]. Thus, the elimination of lung CSCs could increase the therapeutic efficacy of drugs against lung cancer. The CD133 antigen is a putative CSCs marker in solid tumours including lung cancer [Citation10–12]. CD133+ lung cancer cells have been shown to possess stronger potential than CD133– lung cancer cells in self-renewal, proliferation, differentiation and in vivo tumour formation in mice [Citation13,Citation14].
In recent years, targeted drug delivery system has become a hot research topic, since they could improve the targeting efficacy of chemotherapy drugs [Citation15,Citation16]. The combination of ligand–receptor was a classical method for targeted drug delivery [Citation17,Citation18]. Aptamers – oligonucleic acids – have advantages including low immunogenicity, low molecular weight and easy production, making them ideal targeted ligands [Citation19]. The aptamer A15 has been demonstrated to be a promising ligand for targeting CD133+ cells [Citation20]. Given that CD133 is a marker of lung CSCs, we hypothesize that A15–CD133 interaction could mediate effective delivery of drugs to CD133 positive lung CSCs.
Docetaxel (DTX) belongs to microtubule depolymerizing agents, which also includes paclitaxel (PTX). The structure of DTX is similar to that of PTX. Clinically, DTX is used to treat many types of cancers such as breast cancer, ovarian cancer, lung cancer and prostate cancer by inhibiting mitotic progression, cell apoptosis [Citation21]. However, poor water solubility (6–7 µg/mL) remains a critical limitation of DTX. Taxotere (commercially marketed brand) contains dehydrated alcohol (0.395 mg/mL) to improve the solubility of DTX [Citation22]. In addition, multidrug resistance of DTX decreases the accumulation of drug within the cell and increased the repairing mechanism of DNA damage leading to therapeutic efficiency of single agent.
The aim of the present study was to prepare a novel CD133 aptamer modified DTX liposome system and investigate its characteristics, including size distribution, zeta potential, drug entrapment efficiency and morphology. Cellular uptake of the liposomes was assessed by fluorescence microscopy. Finally, an A549 tumour model was constructed and the tumour-targeting potential of the liposome in vivo was examined.
Materials and methods
Materials
DTX was purchased from Haizheng Biopharma Ltd., Co. (ZhaJiang, China). Soybean phosphatidylcholine (SPC), DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000]) and cholesterol (CHOL) were obtained from Sinopharm Chemical Reagent (Shanghai, China). A549 cell was purchased from Gefan Biopharma Ltd., Co. (Shanghai, China). The primers and thiolated CD133 aptamer with a sequence of 5′-SH-CCCUCCUACAUAGGG-3′ were bought from Ruibo Co., Ltd. (Guangzhou, China). All other reagents were obtained from Sinopharm Chemical Reagent (Shanghai, China). Methanol and acetonitrile (chromatographic grade) were obtained from Sigma-Aldrich (St. Louis, MO). Water for HPLC was double distilled and all other reagents were analytical grade.
Preparation of liposomes
The liposomes containing DTX were prepared by the thin-film hydration method. Briefly, a mixture of DTX, SPC, CHOL and DSPE-PEG2000 (The molar ratio of DSPE-PEG2000:CHOL:SPC was 1:5:10, the weight ratio of lipid:DTX was 15:2. Then, the solvent (5 mL chloroform) was evaporated using an rotary evaporator in a round-bottomed flask at 40 °C for about 30 min to obtain a solid film. This film was then flushed with nitrogen for 30 min and stored overnight in a desiccator to remove any traces of chloroform. After that, the thin-film was hydrated by sonication in 5% glucose solution and in water bath for 10 min to produce a suspension of liposomes. The targeted modified DTX liposomes with CD133 aptamers were fabricated by conjugating CD133 aptamers to DTX liposomes by a thiol-maleimide reaction [Citation23,Citation24]. In brief, DTX liposomes were prepared as described above, and then the DTX liposomes suspension was incubated with CD133 aptamers (1% m/m) for 8 h under stirring. After ultrafiltration with PBS, the CD133 aptamers modified DTX liposomes (CD133-DTX LP) were resuspended in PBS for use.
Characterization of liposomes
The morphology of the liposomes was observed using a transmission electron microscope (JEM-1200EX, Tokyo, Japan). The samples were added to the surface of copper grids, and stained with phosphotungstic acid (1%, w/v). The accelerating voltage was of 120 kV. The particle size and zeta potential of the DTX liposomes and CD133-DTX LP were measured by dynamic light scattering (NicompTM380ZLS, Particle Sizing Systems, Santa Barbara, CA). Before analysis, each sample was diluted 20-fold in distilled water (pH 7.4) to obtain the appropriate liposomal concentration. The encapsulation efficiency of DTX in the liposomes was determined by HPLC. Briefly, 0.5 mL of the liposomal sample was first added in Super filter and then centrifuged at 12,000 rpm for 30 min. The amount of ultrafiltrated DTX was measured by HPLC. The encapsulation efficiency of DTX was calculated using the formula: % Drug loading = (Wtotal – W(free drug))/Wliposomes. Where W(free drug) was the analysed weight of free drug, Wtotal was the weight of drug in liposomes; Wliposomes were the analysed weight of liposomes.
DTX content in the samples was analysed by HPLC (Agilent 1100, Santa Clara, CA). The mobile phase consisted of acetonitrile and water (70:30). The chromatographic conditions were as follows: an ultraviolet detector (Agilent 1100) set at 230 nm; a C18 column (Gemini ×5 µm) operated at 30 °C ± 1.0 °C; and a flow rate of 1 mL/min. The injection volume was 20 µL.
In vitro release study
In vitro release of DTX from LP and CD 133 aptamers modified LP was monitored by means of dialysis method. In this study, 1 mL of LP was placed in the dialysis bag and both the borders were sealed with a dialysis clip. The dialysis bag (molecular cut-off of 10 kDa) was incubated in 25 mL of release media (PBS, pH 7.4 containing 1% Tween 80 as a solubilizer). The whole set up was placed in an automated shaker maintained at 100 rpm and 37 °C. At predetermined time intervals, 1 mL sample was collected and replaced with equal amount of fresh media. The released drug was quantified using HPLC method as mentioned above.
Stability study
The particle size, zeta potential and Entrapment efficiency were evaluated at 2–8 °C for 3 months as the chemical and physical stability. The centrifuge tests were also carried out to assess the physical stability of the CD133-DTX LP. The liposomes were centrifuged for 30 min at 650×g in the centrifuge tests.
Haemolytic activity testing
Haemolytic activity was evaluated by determining haemoglobin release from erythrocyte after incubation with different DTX liposome formulations. Briefly, rabbit blood samples were harvested from arteria auricularis into test tubes containing 124 mM sodium citrate (sodium citrate:blood = 1:9, v/v), centrifuged and washed with saline. The obtained red blood cells (RBCs, 1 mL) were diluted with saline to 10 mL. 0.5 mL of the RBC suspension was incubated with 2 mL of liposomes (2.5 mg/mL) at 37 °C with gentle shaking. After 1 h, the samples were centrifuged at 3000 rpm for 5 min. The absorbance (A) of the supernatant was measured by UV–vis spectrophotometry at 545 nm. A negative control was prepared by mixing 0.5 mL of the RBC suspension with 2 mL of saline (0% lysis), using water as a positive control (100% lysis). The absorbance value of positive should be 0.8 ± 0.3, while negative one should be less than 0.03. The haemolytic rates of the samples were calculated as the following equation:
where At represents absorbance value of test sample, Anc and Apc stand for absorption value of negative and positive controls, respectively.
Cellular uptake
A549 cells were grown in RPMI 1640 medium supplemented with 10% (v/v) FBS and 5% antibiotics (100 IU/mL of penicillin G sodium and 100 µg/mL of streptomycin sulphate). The cellular internalization of free DTX, DTX LP and CD133-DTX LP was visualized by confocal microscopy using coumarin-6 as a fluorescent probe. A549 cells were seeded in a cell culture dish at an initial density of 4 × 105 cells per dish. Cells were then incubated with coumarin-6-adsorbed free DTX, DTX LP and CD133-DTX LP (equivalent to 0.1 µg/mL of coumarin-6) for 2 h at 37 °C ± 0.5 °C. Subsequently, cells were washed several times with PBS and fixed with 4% paraformaldehyde for 10 min. Finally, cells were observed under a confocal microscope. For quantitative estimation of DTX uptake, cells were seeded in 24-well plates at a density of 3 × 104 cells. When they reached 70–80% confluence, cells were incubated with coumarin-6-adsorbed free DTX, DTX LP and CD133-DTX LP (equivalent to 0.1 µg/mL of coumarin-6). After 2 h of culture, cells were washed several times with cold PBS. Subsequently, cells were dissolved by addition of Triton X-100 (0.1%). Fluorescence intensities were measured by a multimode microplate reader at an excitation wavelength of 440 nm and an emission wavelength of 520 nm.
Cytotoxicity study
The cytotoxicity of blank LP, free DTX, DTX LP and CD133-DTX LP was evaluated in A549 cells by MTT assay. Briefly, A549 cells were seeded in a 96-well plate at a density of 3–4 × 103 cells per well. After 12 h, different DTX formulations (equivalent to DTX concentrations ranging from 0.002 µg/mL to 20 µg/mL) were added, and plates were incubated for 24 and 48 h. DTX standard solution was prepared with DTX dissolved in ethanol ranging from 0.05 to 2 mg/mL and then 100 times diluted with distilled water. Measurements were taken using a microplate reader.
In vivo imaging study
The CD133-overexpressing tumour xenograft model was established by inoculation of 1 × 107 cells (in 100 µL of serum free medium) into the subcutaneous tissue of the right axilla in live nude mice. When the size of the tumours reached 100–150 mm3, 200 µL of Fluorescent probe Dir-labelled DTX LP and CD133-DTX LP was injected into the tumour-bearing mice via the tail vein. After the mice were anesthetized, whole body fluorescence images were acquired using the IVIS Imaging System (CRI Maestro) at different time points (2, 8 and 10 h). The exposure time was set at 600 ms, and the fluorescence signal was collected at 780 nm. Fluorescence images of dissected organs (brain, heart, liver, spleen, lung, kidney) sacrificed 10 h after intravenous injection were also collected. Meanwhile, the fluorescence images of dissected tumours of mice 10 h after intravenous injection of DTX LP and CD133-DTX LP were also analysed.
In vivo antitumour activity
The study protocol was approved by the Animal Use Committee of Soochow University (Suzhou, China) and was carried out in accordance to with the guideline of experimental animals of Soochow University. To evaluate the antitumour activity of PBS, Free DTX, DTX LP and CD133-DTX LP in vivo, tumour-bearing mice were randomly divided into three groups (eight mice each group). The BALB/c nude mice (aged 6 weeks, weight 20–22 g) had been inoculated subcutaneously with 2 × 109 human lung adenocarcinoma (A549) cells. The treatments were started on the day when the tumour volume reached 100–150 mm3, which was set for day 0. Mice in each group were intravenous administrated free DTX, DTX LP and CD133-DTX LP at DTX dose of 10 mg/kg. Tumour sizes and body weights were recorded every three days. The tumour diameters were measured on alternate days with a vernier calliper in two dimensions. Individual tumour volumes (V) were calculated using the formula: V = (L×W2)/2, where in length (L) is the longest diameter and width (W) is the shortest diameter perpendicular to length.
Statistical analysis
Data were presented as mean ± SD. Statistical analysis was performed using one-way analysis of variance for multiple samples and Student’s t-test for comparing paired sample sets. A p value less than .05 was considered statistically significant and p < .001 was highly significant.
Result and discussion
Characterization of liposomes
In recent years, with the development of tumour molecular biology, more and more researches had indicated that a rare amount of a type of cancer cell existed in tumour tissue, which acted as a stem cell during tumour formation process, with potential of self-renewal, proliferation and differentiation. Although its quantity was small, it played important role during tumour occurrence, development, recurrence and metastasis. Since its many properties were similar to stem cells, those cells were called as tumour stem cells. With a deeper research on tumour stem cells, more and more tumour stem cell markers were found, in which CD133 was a type of tumour stem cell markers that was most widely studied. Some researches confirmed that lung CSCs belonged to CD133 cell subsets. This type of tumour stem cell had sphere formation ability and rapid proliferation and differentiation ability. Thus, CD133 became new target of study on lung cancer treatment. DTX is a potential cytotoxic drug with a strong antimitotic action against broad spectrum of cancers including lung cancer. Its cytotoxic effect is related with its enormous potential to inhibit cell proliferation and cell apoptosis [Citation25–27]. In this study, the liposomes containing DTX and CD133 aptamers were prepared by the thin-film hydration method. The characterization of DTX LP with or not CD133 aptamers modified is shown in . Both DTX LP were found to be in the size of 100–120 nm and that of the larger particle size of CD133-DTX LP as compared to DTX LP could be due to the anchoring of CD 133 aptamers molecule at the surface of LP and hence an increment in size of LP was observed. The TEM photomicrographs are shown in . It was observed from these photomicrographs that all samples of particles were smooth, sub-spherical in shape and aggregated to form small clusters. The zeta potential of particles is often used to characterize the surface charge properties of particles. Compared with the DTX LP, modified LP can achieve higher drug encapsulation efficiency, which may be due to the minimal repulsion between the drug and polymer [Citation28].
Figure 1. The structure of CD133-DTX LP (A) and the transmission electron microscope of DTX LP (B) and CD133-DTX LP (C) (×97,000).
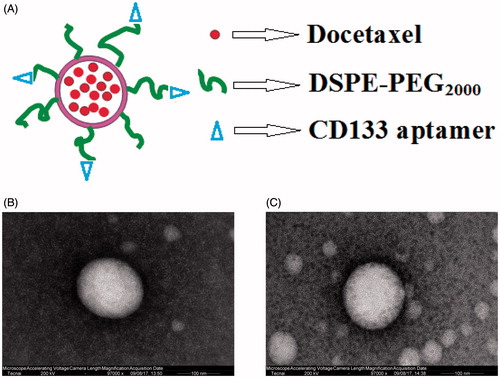
Table 1. The characteristics of different formulations: particle size, entrapment efficiency, polydispersity index and zeta potential (n = 3).
In vitro release study
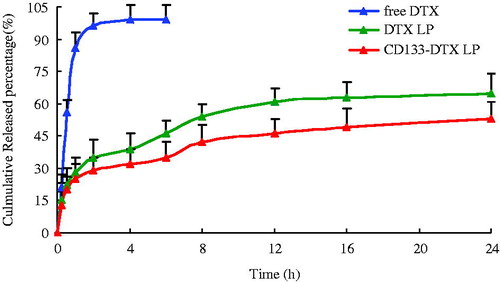
The in vitro release study was conducted in PBS media at 37 °C (). Over time, DTX in liposomes was released much more slowly than free drug. Both DTX LP and CD133-DTX LP exhibited a monophasic release pattern. Throughout the study period, 65% and 60% of DTX released from LP and CD133 modified LP system at the end of 24 h of study period. Noticeably, a relatively slower DTX release profile was observed in CD133-DTX LP due to the presence of CD133 aptamers on the outer surface which might hinder the drug release. To investigate the drug release mechanism, the release data were fit to models representing zero order, first order, Higuchi and Ritger–Peppas equations. Correct determination of the release mechanism depends greatly on the selection and application of a suitable model to the release data. Model fitting of 24 h reveals that in vitro drug-release kinetic model of CD133-DTX LP in PBS fit well with the Higuchi equation: Q = 3.625t1/2 + 0.1623 (r = 0.9991). Thus, it was speculated that the sustained-release property of CD133-DTX LP could enhance cycles absorption of DTX.
Figure 2. The release profile of free DTX (top line), DTX LP (middle line) and CD133-DTX LP (bottom line) (n = 6).
Stability study
CD133-DTX LP exhibited good stability in the period of three months. No significant change in clarity and phase separation was observed in the tests of centrifugation. CD133-DTX LP had a good physical stability. This means that the change process of dispersed lipid in liposomes dispersion from β′ form to stable β form might was very slow. CD133-DTX LP in the formulations was observed with no degradation ().
Table 2. Physical stability of CD133-DTX LP at 2–8 °C (n = 3).
Haemolytic activity testing
Haemolysis study was performed to investigate the potential toxicity after the intravenous injection of DTX LP and CD133-DTX LP in vivo. The leakage of haemoglobin was used to quantitatively compare the membrane-damaging properties of these liposomes. The result showed that the conventional DTX LP showed much higher extents of haemolysis rate than the CD133-DTX LP (9.5% vs 4.6%, p > .05). But there was no significant difference between the two LPs. It is known that, CD133 aptamer has a high degree of segmental flexibility in aqueous solution. Thus, CD133 aptamer-modified is commonly considered to reduce the serious cellular interaction and consequently reduce the damage to RBC. With the improved biocompatibility, the CD133-DTX LP could be further explored for anti-tumour activity, cellular uptake and in vivo effect.
Cellular uptake
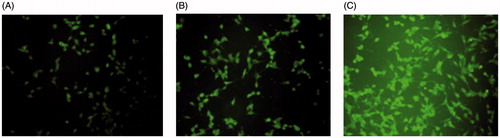
Confocal microscopy was employed to characterize the cellular internalization of free DTX, DTX LP and CD133-DTX LP in A549 cells. As shown in , a strong green fluorescence was observed in the cytoplasmic region after incubation of CD133-DTX LP for 2 h. Cell internalization in A549 cells was higher for CD133-DTX LP than others. Results showed that depending on surface modification, which suggested that cell internalization was changed and more drugs entered the cells successfully. In quantitative cell uptake studies, coumarin-6 on the five formulations was quantified by recovering the drug LPs from cells and measuring their fluorescence (normalized to per mg of the total cellular protein contents). Quantitative results were in agreement with the confocal images (data not shown).
Figure 3. Confocal images of cellular uptake of free DTX (A), DTX LP (B) and CD133-DTX LP (C) by a 549 cells. Incubation time was 2 h.
Cytotoxicity study
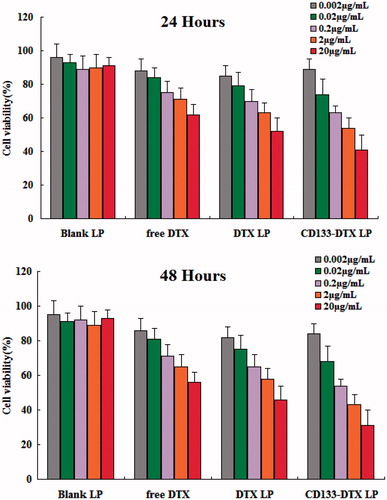
Cell inhibition effect of different DTX formulations on A549 cells were evaluated by MTT assay (). It can be seen that, free DTX, DTX LP, CD133-DTX LP exhibited a time-dependent and dose-dependent cytotoxicity on the cell lines tested. Although DTX is a powerful antitumour drug yet it cannot completely inhibit the cell proliferation. Similarly LP conjugation of DTX did not result in superior effect. On the other hand, CD133 aptamers modified DTX LP significantly decreased cell proliferation and improved the therapeutic efficiency. Surprisingly, CD133-DTX LP showed a superior antiproliferative effect on A549 cells with the possible mechanism is that through the CD133 aptamers mediated, DTX could be delivered to the tumour tissue effectively, and thus play a therapeutic effect.
Figure 4. In vitro cytotoxicity analysis of free DTX, DTX LP and CD133-DTX LP on A549 cell lines. Cell viability assay was performed by MTT assay.
In vivo imaging study
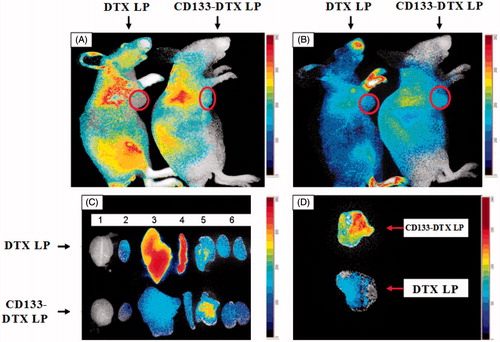
shows that after given near infrared probe Dir 2 h or 8 h, examined by live imager, CD133-DTX LP had stronger fluorescence in nude mouse subcutaneous tumour site than DTX LP. Ten hours after administration, nude mouse was executed, tissues including heart, liver, spleen, lung, kidney, brain and tumour were collected, and found that CD133-DTX LP had stronger fluorescence distribution in tumour tissues than DTX LP. This result indicated that CD133-DTX LP had obvious targeting characteristics on subcutaneous tumour, with potential to passive deliver drugs to tumour tissues. In this research, DTX LP showed relatively high enrichment in liver and spleen (). The reason is possible that after entering animal bodies, liposoluble Dir was separated by haemodilution, and then ingested by liver and spleen, which had plentiful reticular endothelial system. This result also indicated that the advantage of liposome modified by CD133, which could increase the concentration of DTX in tumour sites, reduce its distribution in peripheral organs. As the driving source for tumour growth, tumour stem cells could inhibit tumour tissue inside and helpful to inhibit tumour tissue from the fundamental. In some solid tumour tissue, due to the dense growth of tumour tissue, tumour inside pressure was high with less blood vessels, thus drug delivery system was very difficult to enter tumour tissue inside. This research established tumour model, aimed to simulate liposome distribution inside tumour tissues and its inhibiting ability on solid tumour tissue. The result indicated that CD133-DTX LP had very good tumour targeting ability.
Figure 5. Fluorescence images of subcutaneous A-549 tumour-bearing nude mice after intravenous injection of DTX LP and CD133-DTX LP. The circle, the position of the tumour. (A) 2 h after injection. (B) 8 h after injection. (C) Fluorescence image of dissected organs of mice bearing A-549 tumour sacrificed 10 h after intravenous injection. 1, brain; 2, heart; 3, liver; 4, spleen; 5, lung. (D) Fluorescence image of dissected tumours of mice 10 h after intravenous injection of DTX LP and CD133-DTX LP.
In vivo antitumour activity
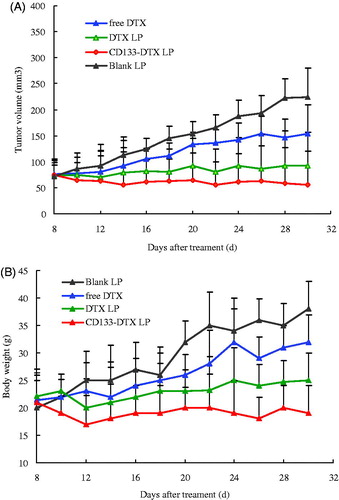
Tumour-bearing mice models were used for the evaluation of the in vivo anti-tumour efficacy of PBS, free DTX, DTX LP and CD133-DTX LP. As shown in , tumour volume of PBS group increased rapidly, while the chemotherapy groups were effective in tumour regression. Notably, the maximum antitumour effect was observed in mice treated with CD133-DTX LP, indicating that this combination therapy produced significant effective inhibition of tumour growth (p < .05). An analysis of body weight variations generally defined the safety of the different therapy regimens (). These findings indicated the CD133-DTX LP showed a significant antitumour activity in A549 tumour mice, with a very low systemic toxicity.
Figure 6. (A) A549 xenograft tumour growth inhibition by DTX in different formulations. (B) Animal body weights. The body weights of treated animals were continuously monitored to investigate systemic cytotoxicity of DTX in different formulations. Data = mean ± SD (n = 8).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Parkin DM, Bray F, Ferlay J, et al. Global Cancer Statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
- Rosell R, Felip E, Garcia-Campelo R, et al. The biology of non-small-cell lung cancer: identifying new targets for rational therapy. Lung Cancer. 2004;46:135–148.
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132.
- Harper E, Dang WB, Lapidus RG, et al. Enhanced efficacy of a novel controlled release paclitaxel formulation (PACLIMER delivery system) for local-regional therapy of lung cancer tumor nodules in mice. Clin Cancer Res. 1999;5:4242–4248.
- Hirsch FR, Suda K, Wiens J, et al. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 2016;388:1012–1124.
- Popper HH. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016;35:75–91.
- Burris HA. Shortcomings of current therapies for non-small-cell lung cancer: unmet medical needs. Oncogene. 2009;28:S4–S13.
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261.
- Gao J, Li W, Guo Y, et al. Nanomedicine strategies for sustained, controlled and targeted treatment of cancer stem cells. Nanomedicine (Lond). 2016;11:3261–3282.
- Xie F, Xu W, Yin C, et al. Nanomedicine strategies for sustained, controlled, and targeted treatment of cancer stem cells of the digestive system. WJGO. 2016;8:735–744.
- Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344.
- Lundin A, Driscoll B. Lung cancer stem cells: progress and prospects. Cancer Lett. 2013;338:89–93.
- Bertolini G, Roz L, Perego P, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286.
- Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514.
- Akbal Ö, Erdal E, Vural T, et al. Comparison of protein- and polysaccharide-based nanoparticles for cancer therapy: synthesis, characterization, drug release, and interaction with a breast cancer cell line. Artif Cells Nanomed Biotechnol. 2017;45:193–203.
- Park BW, Zhuang J, Yasa O, et al. Multifunctional bacteria-driven microswimmers for targeted active drug delivery. ACS Nano. 2017;11:8910–8923.
- Li S, Wang XP. In vitro and in vivo evaluation of novel NGR-modified liposomes containing brucine. IJN. 2017;12:5797–5804.
- Johnsen KB, Burkhart A, Melander F, et al. Targeting transferrin receptors at the blood–brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci Rep. 2017;7:10396.
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550.
- Shigdar S, Qiao L, Zhou SF, et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84–95.
- Du N, Song L-P, Li X-S, et al. Novel pH-sensitive nanoformulated docetaxel as a potential therapeutic strategy for the treatment of cholangiocarcinoma. J Nanobiotechnol. 2015;13:17.
- Blumenthal GM, Scher NS, Cortazar P, et al. First FDA approval of dual anti-HER2 regimen: pertuzumab in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer. Clin Cancer Res. 2013;19:4911–4916.
- Gao J, Liu W, Xia Y, et al. The promotion of siRNA delivery to breast cancer overexpressing epidermal growth factor receptor through anti-EGFR antibody conjugation by immunoliposomes. Biomaterials. 2011;32:3459–3470.
- Gao J, Sun J, Li H, et al. Lyophilized HER2-specific PEGylated immunoliposomes for active siRNA gene silencing. Biomaterials. 2010;31:2655–2664.
- Chowdhury S, Burbridge S, Harper PG. Chemotherapy for the treatment of hormone-refractory prostate cancer. Int J Clin Pract. 2007;61:2064–2070.
- Fulton B, Spencer CM. Docetaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of metastatic breast cancer. Drugs. 1996;51:1075–1092.
- Azargoon A, Negahdari B. Lung regeneration using amniotic fluid mesenchymal stem cells. Artif Cells Nanomed Biotechnol. 2017; 4:1–5.
- Islami M, Mortazavi Y, Soleimani M, et al. In vitro expansion of CD 133+ cells derived from umbilical cord blood in poly-l-lactic acid (PLLA) scaffold coated with fibronectin and collagen. Artif Cells Nanomed Biotechnol. 2017;6:1–9.