
Abstract
Skin is the largest external organ in the human body but its use for therapeutic purposes has been minimal. Stratum corneum residing on the uppermost layer of the skin provides a tough barrier to transport the drugs across the skin. Very small group of drugs sharing Lipinski properties, i.e. drugs having molecular weight not larger than 500 Da, having high lipophilicity and optimum polarity are fortunate enough to be used on skin therapeutics. But, at a time where modern therapeutics is slowly shifting from use of small molecular drugs towards the use of macromolecular therapeutic agents such as peptides, proteins and nucleotides in origin, skin therapeutics need to be evolved accordingly to cater the delivery of these agents. Physical technologies like iontophoresis, laser ablation, micro-needles and ultrasound, etc. have been introduced to enhance skin permeability. But their success is limited due to their complex working mechanisms and involvement of certain irreversible skin damage in some or other way. This review therefore explores the delivery strategies for transport of mainly peptide and protein drugs that do not involve any injuries (non-invasive) to the skin termed as passive delivery techniques. Chemical enhancers, nanocarriers, certain biological peptides and miscellaneous approaches like prodrugs are also thoroughly reviewed for their applications in protein delivery.
Introduction: transdermal route and protein delivery
Transdermal drug delivery renders a beneficial mode of drug administration over oral and parenteral application [Citation1]. They have the tendency to achieve steady-state drug levels, bypass the hepatic first-pass metabolism, increase patient compliance and reduce gastrointestinal (GI) adverse effects [Citation2]. However, it was not until the latter half of the twentieth century that the skin was identified as a route for systemic drug delivery. It was only in 1979 the first transdermal system for systemic delivery was approved in the USA, a three-day transdermal patch for the treatment of motion sickness. A decade later, Nicotine patches have gained enormous popularity as an alternative form of therapeutics in medicine and public in general. At present, there are about 17 drugs delivered via this route and net market share for transdermal therapeutics is currently projected to be worth US $32 billion only, having grown from $20.5 billion in 2010 [Citation3]. But considering the rate of NME (new molecular entities) approvals per year, transdermal drugs lag (0.1 NMEs/year) compared with topical drugs (0.9 NMEs/year) and pharmaceutical innovation in general (20–40 NMEs/year) [Citation2]. Therapeutic agents currently marketed as transdermal drug products share the common physicochemical properties labelled as Lipinski rules, i.e. (a) modest molecular weight (MW 400–500 Da), (b) a balanced lipophilicity [log(octanol–water partition coefficient), log P, ideally around 1–3] and (c) an optimum solubility both in oil and in water (considering that TDD necessitates crossing the lipophilic stratum corneum (SC) barrier as well as absorption into the systemic circulation). In addition, drug molecules should be extremely potent in nature [Citation4]. In the past 30 years after the first launch of recombinant human insulin Humulin, biopharmaceutical drugs – such as peptides, enzymes, monoclonal antibodies, recombinant protein drugs and antibody–drug conjugates – have revolutionized the pharmaceutical industry [Citation1]. After that, a sum total of 91 protein based drugs manufactured by recombinant technology have been approved by the FDA [Citation5]. Specificity attributed by the complex structural orientation of proteins and potency has been the cornerstone of protein based therapeutics as compared to small molecule drugs [Citation1,Citation6]. However, stability related matters, complexity of their nature lend the proteins difficult for delivery. The general route of administration for protein pharmaceuticals to date is parenteral (intravenous/subcutaneous). However, because majority of the proteins possess short half-lives, this route poses disadvantage of the necessitated repeated drug administrations and poor patient compliance. Few other routes like the oral, pulmonary and nasal routes have also been explored as alternatives and some products of the same are available in the market. However, limitations like GI degradation, low bioavailability and local irritation persist [Citation7]. In such scenario, skin could be the potential alternative for administration of protein drugs across the skin as it bypasses the first pass metabolism, offers prolonged release of drug and exhibits minimal proteolytic activity compared to other routes [Citation6]. shows physical techniques of transdermal drug delivery.
Figure 1. Physical techniques of transdermal drug delivery.
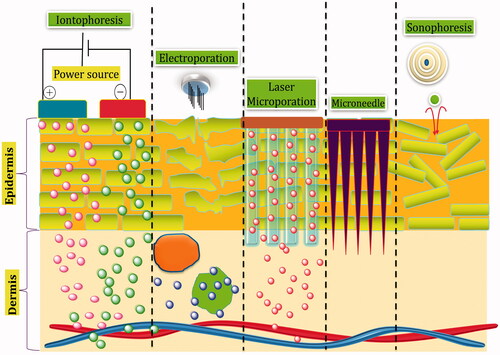
The protein drugs are large in size, have higher MW (average weight of human protein being 53 kDa) and hydrophilic in nature. These properties are antagonistic to Lipinski rules proposed for efficient transdermal delivery, thus suggesting negligible passive diffusion of proteins across the skin. Various transdermal technologies using physical and chemical methods have been introduced in last decade to augment delivery of proteins through the skin such as use of chemical enhancers, iontophoresis, micro-needles, sonophoresis, laser ablation, thermal ablation, radiofrequency ablation, jet injectors, microwaves and electroporation [Citation8]. Except chemical enhancers, all of them consist of mechanical step requiring force in some or other way to mediate permeation of proteins underneath the skin. Iontophoresis and electroporation are the techniques employing electrical assistance which lead to an enhanced drug delivery via skin on application of electric current. With the treatment of low frequency ultrasound (20–100 kHz), sonophoresis enhances the skin permeability facilitating the transport of large molecules including hydrophilic proteins. Microporation techniques involve disruption of SC in a temporary and reversible manner using heat (thermal ablation), radioactive source (radioactive ablation), laser source (laser ablation) or micro-needles. Microwaves use high intensity electromagnetic radiation of 0.3–300 GHz for certain period of time to exert its permeation effect through thermal or non-thermal interaction with skin [Citation9]. Jet injector uses high velocities for bombarding the skin and abrading to create superficial micro-pathways in the skin for delivery of powder and liquid formulations [Citation10,Citation11]. Most of the products using above-mentioned technologies are not currently available for purchase and have been withdrawn from the market by their developers or sellers. Some products simply did not sell in sufficient numbers to warrant their place on the market and others were deemed as possibly unsafe and withdrawn. This experience illustrates that the confluence of a drug and its delivery system, particularly one that involves complex technology described above is never going to be an easy one. Transdermal therapy is widely known for its patient compliance, but when coupled with these complex techniques involving sophistication in design and delivery methods, loses its essence of being the most convenient route. Also both the device and drug must meet stringent safety and performance criteria laid out by FDA which doubts the easy transfer of these technologies into market [Citation2,Citation3]. This review, therefore, focuses on simpler techniques which have the capability to circumvent the inherent nature of skin barrier and deliver the therapeutic amount of protein based drugs without involving any complex physical or mechanical devices.
Stratum corneum and its lipid architecture
Skin acts as a barrier for diffusion of substances through it and the major barrier for most substances resides in the SC, outermost layer of the skin. The SC consists of flat, non-nucleated; keratin enriched dead cells, corneocytes surrounded by intercellular non-polar lipid domains. Interconnecting the corneocytes are protein structures, referred to as desmosomes [Citation12,Citation13]. Corneocyte wall is a very dense, cross-linked protein structure that reduces absorption of drugs into the cells [Citation14]. The intercellular lipid matrix forms a continuous pathway from the skin surface to the viable skin tissues creating the principle path of entry for many chemicals. Therefore, it is crucial to understand the nature of the extracellular lipid matrix of the SC to deal with the permeability of human skin () [Citation15].
Figure 2. Different penetration pathways across the skin (the upper right corner shows the channel formed by intercellular lipid matrix).
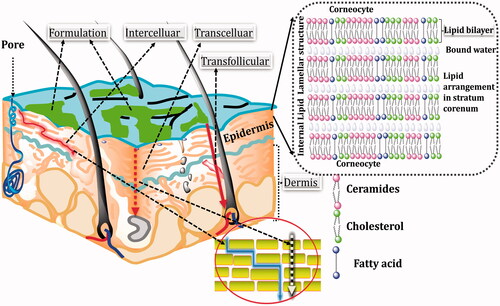
Human SC lipids consist of free fatty acids (FFAs), ceramides (CERs) and cholesterol (CHOL) in an approximately equimolar ratio. FFAs in humans are predominantly saturated and consists of chain lengths up to 36 carbon atoms, with C12, C18, C24 and C26 being the most prominent FFAs. In addition to the saturated FFAs, minimal amount of mono-unsaturated FFAs (MUFA), poly unsaturated fatty acids (PUFAs) and hydroxyl FFAs are found. Unlike FFAs, CERs consist of two carbon chains, viz. one fatty acid (acyl) amide connected to sphingoid base. The acyl chain of CERs differs from C14 to C32 and that of sphingoid base constitutes between C14 and C28. In addition, each acyl and sphingoid base has an additional functional group. The variety of chain lengths and head group structures thus results in the existence of over 400 different CER species. Next to CHOL, CERs and FFAs, SC also contains other lipid classes, such as the glucosyl CERs, precursor of CERs, and CHOL sulphate having significant part in desquamation process [Citation16]. The arrangement of lipid matrix in SC can be understood by lateral organization (i.e. the molecular packing of lipids in lamellar plane generally parallel to the SC surface) and lamellar organization (i.e. the symmetry and the repeated distance perpendicular to the SC surface of lipids). Lamellar organization of SC consists of stacks of lamellae of so-called broad-narrow-broad arrangement having regular repetitive units with a repeated distance of 13 nm particularly referred to as long periodicity phase (LPP). Besides a 13 nm lamellar phase, a second lamellar phase has also been detected having periodicity approximately 6 nm and is therefore called as the short periodicity phase (SPP). The LPP is found between the large, flat surfaces of adjacent corneocytes and the SPP – close to their edges. Lateral lipids in the intercellular matrix can adopt three varieties of packing arrangements which differ in their rotational and translational motilities. In the most densely packed, orthorhombic (OR) phase, the lipid chains adopt an all-trans conformation and are organized in a rectangular crystalline lattice with no rotational or translational mobility. In the hexagonal (HEX) phase, the all-trans lipid chains have some rotational mobility along their long axis, but their translational mobility is restricted whereas in the liquid-crystalline (LIQ) phase, the chains have both high rotational and high translational mobility. All these three phases co-exist in healthy human SC, with a notable prevalence of the OR phase [Citation15,Citation17].
Role of lipids in the permeability barrier
LPP examined in all species until now, has suggested that the presence of this phase plays an important role in skin barrier function. In the SC of patients suffering from several diseases characterized by very high skin permeability (atopic dermatitis, lamellar ichthyosis, psoriasis, Netherton syndrome and Chanarin–Dorfman disease), the periodicity of the LPP is decreased and its general appearance is significantly altered [Citation18]. During investigations on the penetration of ethyl para-amino benzoic acid and benzoic acid across model SC membranes, the fluxes of these chemicals were found to elevate in the absence of LPP which supports the positive role of LPP in permeability across the skin. The lateral lipid organization within the lamellae strongly influences the SC permeability for water. The flux of water across human skin closely correlates with the type of solid phases present in the SC: the higher the OR content, the lower the flux of water [Citation13,Citation15,Citation17]. The permeability through the skin therefore is the interplay of arrangement of lipids in lamellar and lateral layer. Absence of LPP favours high permeability whereas prevalence of OR arrangement in lateral layer reduces the permeation. Therefore, any approach for enhanced permeability through skin should be directed towards achieving LIQ arrangement of lipids laterally as opposed to OR and perturbing the LPP phase in lamellar symmetry.
Methods of passive protein delivery through skin
Use of various mechanical methods for protein delivery notably iontophoresis, thermal ablation, ultrasonication, laser methods involves irreversible skin damage. Hence, non-mechanical passive methods are reviewed below as shown in for their obvious advantages of simplicity in use, patient compliance and skin reversibility after application.
Figure 3. Strategies for passive delivery of proteins.
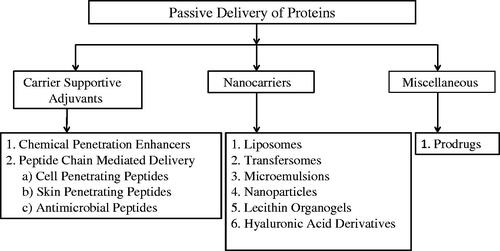
Carrier supportive adjuvants (CSAs)
Carrier supportive adjuvants are auxiliary chemical moieties used in association with carriers like liposomes, microemulsions, gels for penetration enhancement during transdermal delivery. Since they are required to be incorporated in some other mediums for their functioning, they are classified broadly as chemical penetration enhancers (CPEs) and peptide chain mediated delivery under CSAs.
Chemical penetration enhancers
Chemical penetration enhancers are those classes of auxiliary chemical moieties when incorporated along with the principal penetrant moieties, i.e. drug molecules, protein formulations, etc. assist in the penetration of these principal moieties across the skin by altering the permeability barrier of skin. The exact mechanism of CPEs is vaguely understood but it has been assumed that the association of CPEs with lipids of SC helps in creating a perturbed microenvironment through which a drug can freely diffuse. Ethanol, propylene glycol, oleic acid, transcutol, dimethyl sulphoxide surfactants and azones, etc. are commonly used penetration enhancers ().
Table 1. Various classes of CPEs and their mechanism of action.
Bhatia and Singh reported that pretreatment with 5% terpenes (thymol, cineole, carvone and limonene) in combination with the ethanol significantly increased the passive transport of LHRH through porcine skin. The flux obtained using 5% limonene was 6.60 ± 1.20 [(nmol/cm2/h) × 106] and enhancement factor was found to be 4.7 times passive transport, highest among other used terpenes [Citation25]. Similar findings on LHRH permeability were reported by Smyth et al. on human epidermal membrane (HEM). LHRH penetration was increased by 3.5 times over passive permeability using oleic acid, propylene glycol as penetration enhancer [Citation26]. Under the influence of CPEs, the in vitro cutaneous permeation study of IFNα into human skin was studied by Moghadam et al. They studied the effect of three groups of CPEs; solvents [ethanol, propylene glycol, diethylene glycol monoethyl ether (DEGEE) (transcutol), oleic acid], terpenes [menthol, nerol, camphor, methyl salicylate] and surfactants [Tween 80, SDS, benzalkonium chloride, polyoxyl 40 hydrogenated castor oil (Cremophor RH40), didecyldimethylammonium bromide (DDAB), didecyltrimethylammonium bromide (DTAB)]. The highest permeation across the skin was observed with surfactants, followed by the terpene and solvent CPEs. The permeation enhancement was found to be more for Tween 80 and Cremophor RH40 with 6- and 3.7-fold enhancement compared with the IFNα solution without any CPE, which corresponds to 0.82% and 0.52% of IFNα dose, respectively. Cationic surfactants did not appear to promote permeation of IFNα to a greater extent [Citation27]. Permeability enhancement of cholecystokinins (CCK-8) through the use of CPEs was evidenced by the fact that their permeability coefficient was 2.18 times greater by the use of ethanol and 3.93 times higher by employing 10% oleic acid and ethanol in combination when compared to CPE untreated skin. [Citation28]. CPEs are efficacious in breaching the barrier property of skin to improve the delivery of drug molecules in therapeutic amount but their safety is a potential concern. Large number of CPEs (such as azone derivatives, fatty acids, alcohols, esters, sulphoxides, pyrrolidones, glycols, surfactants and terpenes) are small molecules (500 Da) that can penetrate the skin in significant quantities and can cause skin irritation, cytotoxicity or irreversibly change the barrier properties of the skin. Therefore, a delicate balance should be established between transport enhancement and skin irritation in case of CPEs [Citation29].
Peptide chain mediated delivery
Some of the peptides possess good skin penetration enhancing properties or act as carriers for drug delivery via transdermal route. They possess advantages like easy usage, diversity and capability of targeting specific cells within the skin, and feasibility of conjugation with drugs [Citation30]. Apart from its enhancement of skin permeability, they are also interrupting barrier properties, for the improved delivery of therapeutic agents or cargoes across the skin. At present, three pathways exist for the transport of drugs across the skin; intracellular pathway, intercellular lipid pathway and the trans-appendageal pathway. The intercellular lipid pathway has been widely used for the delivery of cargoes either in free form or in a conjugated form [Citation31]. describes various types of transport mediated by peptides along with their probable mechanisms of permeation.
Figure 4. Peptide mediated transport mechanisms for transdermal permeation.
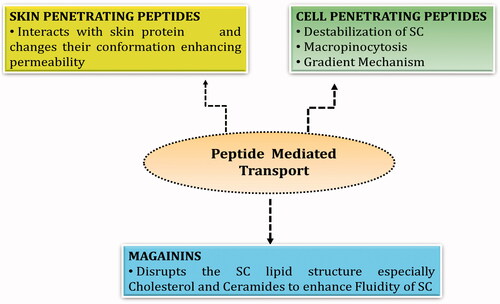
Cell penetrating peptides
CPPs are amphiphilic peptides consisting up to 30 amino acids derived from natural or unnatural protein or chimeric sequences, which can be internalized by cells through energy independent mechanisms with or without the aid of receptors. All known CPPs are net positively charged at physiological pH, and incorporate approximately 17–100% of positively charged amino acids [Citation32,Citation33]. Most CPPs are not specific to any tissues or cells instead they depend on the positive charge of amino acid sequences at physiological pH (primarily arginine and lysine) and electrostatic interactions with negatively charged glycoproteins of cell surface [Citation34]. CPPs have inherent property to aid in translocation of agents through plasma membrane which have been widely exploited for cellular uptake and membrane interaction studies. In addition, they have unique feature of structural modifications for enhancing delivery of macromolecular agents [Citation35]. CPPs alone and their cargoes are relatively safe at the concentration used for delivery of macromolecules due to their low levels of cytotoxicity.
Types of cell penetrating peptide
CPPs are divided into different classes based on the criteria for division. On the basis of association with cargoes, they are divided into two types: the one requiring chemical linkage with cargo other being the one forming stable, non-covalent complexes. On structural basis, they are divided into either polycationic or amphipathic classes [Citation32]. shows some of the most widely explored CPPs for delivery into the cells.
Table 2. Widely explored CPPs.
Cell penetrating peptide in transdermal delivery of proteins
Cell penetrating peptides are commonly utilized for the delivery of drugs which causes problem for transportation of drug across cell membrane. CPPs have been used to improve delivery of cargo that varies greatly in size and nature (small molecules, oligonucleotide, plasmid DNA, peptide, protein, nanoparticle, lipid-based formulation, virus and quantum dots). They are most commonly employed for the delivery of oligopeptide/protein and nucleic acids or analogues [Citation32]. Jain et al. reported enhanced corneal permeation of natamycin upon conjugation with Tat2 (trans activator of transcription) in efficient management of fungal keratitis [Citation43]. The skin permeability of diclofenac sodium and celecoxib was enhanced by six-fold and eight-fold, respectively, employing cell penetrating peptide HIV-TAT in cubic phase [Citation44]. Tat-CAT and 9Arg-CAT fusion proteins, i.e. catalase enzyme was conjugated with Tat and nine arginine residues, respectively. When these were sprayed on animal skin they penetrated the epidermis as well as the dermis layer with good therapeutic efficiency against antioxidant related disorders [Citation45]. Hou and Kong studied transdermal delivery of proteins which were non-covalently associated with arginine. An enhanced intracellular delivery of peptide was observed in the presence of chemical enhancers [Citation46]. OVA257–264 is a protein derived antigen which was linked to Antennapedia onto mice skin, when administered topically resulted in enhanced delivery through the skin and augmented anti-tumour immunity of the antigen. This approach provided a technical and theoretical basis for the further development of novel peptide-based cancer vaccines using CPP [Citation47]. Melittin, a cationic antibacterial protein resembling to CPP properties, is an efficient penetrant for the abdominal skin corneum to reach the dermis. It possesses the capability treating non-melanoma cancer and skin infection [Citation18]. Gennari et al. synthesized heptapeptide (DRTTLTN) and studied its permeation effects for unfractioned heparin (UFH) on human SC. However, the co-administration of DRTTLTN with UFH was found to be ineffective in enhancing skin penetration because of high affinity of UFH to keratins. When the peptide was conjugated with N-3-(dimethylamino) propyl) – N′-ethylcarbodiimide hydrochloride and sodium N-hydroxysulfosuccinimide, flux increased by 24–36 times [Citation48]. In another study, de Cogan et al. evaluated the efficacy of anti-VEGF agents via topical route for the treatment of choroidal neovascularization (CNV) using novel cell-penetrating peptides (CPPs) against an intravitreal (ivit) injection of the same. CPP in combination with anti-VEGF agent showed clearance from the retina within 24 h. In vivo studies revealed that the efficacy of topically delivered anti-VEGF with CPP was more or less similar to i.v. injection of anti-VEGF in reducing the areas of CNV [Citation49].
Some CPP based formulations were successful to even enter in clinical trials. One of such example is cyclosporine octaarginine (Psorban by Cell Gate International) for the treatment of Psoriasis by transdermal delivery of cyclosporine A. It has successfully entered phase II clinical trials [Citation50].
Mechanism of penetration of CPP across skin
The entry of CPP or its cargo complex into the cell is the widely studied area compared to transport of CPP across the skin but universal mechanism of transport across cell membrane applied to all classes of CPP is yet to be agreed upon [Citation33]. However, it has been suggested that the transport could be affected by variety of factors, including the nature of the cargo (such as size and charge), the properties of CPP (such as molecule length, charge delocalization, hydrophobicity and other physicochemical parameters), the cell line and the CPP concentration. Non-endocytic or energy-independent pathway and the endocytic pathway among others are the two important pathways proposed for CPP penetration inside the cells. Both mechanisms involve (i) membrane interaction, (ii) membrane permeation and (iii) release of CPP into the cytosol after the entry procedure. Moreover, models such as inverted micelle formation, pore-like structure formation, the carpet-like model and the membrane thinning model are proposed to explain their penetrative nature [Citation47]. The membrane composition and intercellular lipid domain of SC is different from the cellular membrane in terms of lipid composition, lipid/protein ratio and water content. This ipso facto indicates the existence of a varying transport mechanism from the normal cell membrane [Citation51]. Since the outermost layer of the skin is composed of non-viable cells, the possibility of endocytosis can be easily ruled out despite the fact these dead cells form a metabolically active environment. Interactions between lipids and CPPs may also play important role in their transport across the SC. CPPs enable to transport their cargoes across the skin by destabilizing SC and increasing permeability. For example, poly-l-arginine was reported to increase the permeability of tight junctions of the nasal epithelium and facilitate the transport of a dextran. This effect was initiated by interaction of positively charged polyarginine with negatively charged cellular lipids [Citation33,Citation52]. Macropinocytosis is another possible transport mechanism, by which the CPPs pass through the mammalian cells. It has been shown that the macropinocytosis and actin reorganization are both involved in the cellular entry and transdermal delivery of CPP/cargo complexes. Nevertheless there is quite less likelihood of involvement of this mechanism of transport in the penetration of non-covalently attached CPP/cargo complexes. Finally, the gradient factor might be involved as a driving force in CPPs penetration into different layers of the skin. The exact mechanism of CPP transport through skin layers is not fully understood, and further studies are required to identify mechanism(s) responsible for the CPP-mediated transport from nonviable to viable layers of the skin [Citation47]. Di Pisa et al. studied the translocation mechanism of CPP using artificial membrane. The primary step of transport may be viewed as a concentration step from the extra vesicular medium to the membrane surface. These interactions may induce folding and/or an oligomerization of the CPP mediated by the presence of phospholipids. In response to the interaction with the CPP, the lipid bilayer may be profoundly disturbed. Membrane symmetry can be additionally perturbed. Following adsorption and peptide–lipid interaction at the water/membrane interface, CPPs can also insert into the membrane. The nature of events shown here can closely resemble in case of CPP applied to the skin. Further studies should be carried out to validate this sequence of events taking place in skin [Citation53].
Skin penetrating peptides
Small peptides (1000–1500 Da) were introduced a decade ago, labelled as skin penetrating peptides (SPPs) as a safer alternative for improved transport of therapeutic molecules both small and large in nature across the skin [Citation54]. Hsu and Mitragotri extensively used this technique and provided the first evidence that SPPs selected by phage display are able to enhance the penetration of macromolecules through human skin and favour the partition of nanocarrier within the epidermis [Citation55]. Skin presents itself as a formidable barrier and does not allow the penetration of molecules greater than 500 Da comfortably, but the very fact SPPs being 1000–1500 Da acts as an adjuvant for enhanced delivery of macromolecular drugs is ironic. However, it is a potential area for passive transport of protein and peptide drugs that is continuously evolving over a period of time [Citation31]. Chen et al. studied the transdermal permeation of large polysaccharide hyaluronic acid (HA MW: 200–325 kDa) conjugating it with phospholipid and formulating it as ethosomal carrier, SPACE-ethosomal system (SES). In vivo, this system showed enhanced delivery of HA both in vitro performed in human skin and in vivo using SKH1 mice. The SPACE-ethosomal delivery system therefore provides a formulation for topical delivery of macromolecules that are otherwise difficult to deliver into the skin [Citation56]. Additionally, they studied the effect of physical mixture of SPACE-peptide (SP), a class of SPP, and cyclosporin A in an aqueous ethanolic solution for transdermal absorption profile. When hydroethanolic solution was administered in vitro in porcine skin at an amount consisting 5 mg/ml of cyclosporin and 50 g/ml of free SP encouraging results were observed. Approximately, 30% of cyclosporine got delivered and there was an accumulation in greater amount in viable epidermis, i.e. nine times as compared to the control group (hydroethanolic solution without SP). In vivo studies conducted using SKH1 hairless mice further corroborated the results observed during in vitro studies [Citation57].
Mechanism of penetration of SPPs
SPPs did not alter the skin lipid barrier or fluidization as measured by skin resistance, transepidermal water loss (TEWL) and Fourier transform infrared (FTIR) spectroscopic analysis. The lack of a direct effect of SPPs on SC lipids suggests that SPPs do not mediate permeation enhancement through classical intercellular lipoidal pathways. In contrast, SPPs enhanced skin penetration via a transcellular pathway, enhancing its partitioning into keratin-rich corneocytes through concurrent binding of SPP with keratin [Citation54]. Using cyclosporine A as a model protein, Kumar et al. demonstrated the transport mechanism of SPP via transcellular path. For a drug to get transferred transcellularly, it has to pass through hydrophilic keratin packed corneocytes surrounded by multiple hydrophobic lipid layers. Therefore, it necessitates the series of partitioning steps, i.e. from lipid to protein and protein to lipid followed by diffusion from the respective binding sites. Peptides and proteins through their hydrophobic domain have affinity towards the hydrophobic lipid layers surrounding the corneocytes but are unable to partition themselves into hydrophilic corneocytes. SPACE peptide there acts as a mediator and aids the partitioning of proteins into keratin rich corneocytes, thereby helping them to permeate across the skin. The very fact that binding of SPACE peptide with the keratin most effectively correlated the amount of drug transported across the skin rather than other properties of SPACE peptide like peptide surface charge, hydrophilicity consolidates the proposed mechanism [Citation29].
Antimicrobial peptide magainins
Magainin is a 23-amino-acid-long antimicrobial peptide (GIGKFLHSAKKFGKAFVG EIMNS) isolated from the skin of the African frog (Xenopus laevis) having a net +4 charge that binds to negatively charged phospholipid membranes with the aid of electrostatic interactions. Owing to its ability to form pores in the bacterial cell membranes and permeabilize the lipid bilayers, it is also called as pore-forming peptide. The size of pores formed by magainins in lipid bilayers is estimated to be approximately 1 nm diameter [Citation58].
Taking into consideration the ability of magainin to interact with the lipid membranes, its potential utility as a skin penetration enhancer was evaluated by Kim et al. They demonstrated for the first time the use of a pore-forming peptide magainins to increase skin permeability. Magainins alone were incapable of enhancing the transport across the skin. They require the use of surfactants to accompany them for optimal transport. Magainins along with surfactant NLS ethanol increased skin permeability of fluorescein by 47-fold compared to only 15-fold when surfactant was used alone. Confocal microscopy further supported the results revealing the extensive presence into SC when administered in combination with surfactant than when used alone [Citation59]. Additionally, they described the conditions to improve skin permeability by optimizing the pretreatment time and concentration of magainin exposure. They found that skin permeability increased with increasing pretreatment time. Skin permeability also increased with increasing magainin concentration up to 1 mM, but was reduced at a magainin concentration of 2 mM. The permeation of fluorescein (332 Da) was observed up to 35-fold whereas formulation did not enhance skin permeability to larger molecules, such as calcein (623 Da) and dextran (3000 Da) [Citation60]. They also studied the effect of charge of magainins on the penetration of both cationic and anionic drugs taking granisetron and fluorescein as model drug, respectively. The permeation of negatively charged fluorescein decreased as the positive charge of magainins was increased by reducing the pH. The positive charge on magainins was reduced and ultimately reverted to negative charge which caused repulsive interaction with similarly charge fluorescein inhibiting the transport. The transport of positively charged granisetron increased as the positive charge of magainins was decreased by enhancing pH. The charge on magainins was reduced by enhancing pH which aided in the permeation of positively charged granisetron but reducing pH created the positive charge on magainins, thus reducing the transport owing to the repulsive forces acting between the drug and the protein [Citation61].
Mechanism of penetration of magainin
Analysis by multi-photon microscopy, FTIR, XRD and DSC suggested that magainin increases skin permeability by disrupting SC lipid structure, especially CERs and CHOL, thereby enhancing the fluidity of SC [Citation59]. Magainin peptide could be a potential agent for the penetration of small molecules (peptides here) when used in combination with other chemical skin penetration enhancers like surfactants. However, careful consideration of the amino acid composition, its interaction with the charged drug, the pretreatment conditions and time should be done for effective penetration on skin.
Performance comparison of CPEs versus peptide mediated delivery
CPEs are excellent adjuvants for enhancing the flux of drugs across skin. LHRH, IFNα, CCK-8 are some representative examples where CPEs permeated approximately 3–6 times more amount than when drugs were used alone. Oleic acids, terpenes, azones, fatty acid esters have shown promising results among other CPEs. Additionally, various penetration enhancers can be combined in different ratios or can be associated with other active methods like iontophoresis, ultrasound, microneedles for maximizing the penetrative effect. In spite of all these benefits, the use of CPEs is limited by concerns of potential irritation, toxicity and health issues related to skin. Besides, their role in transdermal delivery of large hydrophilic molecules has always been on scrutiny. Recent progress in high throughput screening to discover more potent options or application of design principles may overcome these issues; nevertheless role of CPEs in delivery of large hydrophilic molecules will always be a challenging task [Citation29,Citation62].
Peptide mediated delivery through CPPs, SPPs and magainins are alternative approaches that address the shortcomings of CPEs. Their effects are observed using non-toxic submicromolar range which drastically reduces the toxicity issues predominant in CPEs. Additionally, these peptide carriers are finally degraded into innocuous amino acids in body further mitigating their harmful effects. More importantly, these peptide mediated techniques can transport the large hydrophilic molecules across the skin easily like proteins, peptides, even nucleic acids and siRNA. UFH, VEGF, cyclosporine transport mediated by these carriers in comparable therapeutic range is a testimony of their ability. Peptide mediated delivery can be applied on various different ways. They can be administered simply by preparing a physical mixture having non-covalent interactions or can be conjugated with drug components during delivery or can also be functionalized into nanocarriers like liposomes, nanoparticles and microemulsions [Citation63,Citation64].
Nanocarriers
Innovative nanocarriers have been designed to transfer molecules across the deep layers of the skin. The degree of penetration enhancement through some nanocarriers has been found to be much greater than that of CPEs [Citation65]. Nanocarriers commonly employed for protein delivery studies are described below:
Liposomes
Liposomes were the original vesicular nanocarriers to be explored for the improved topical effect as compared to conventional topical agents like creams, gels, ointments. Mezei and Gulasekharam in 1980 found the liposomal triamcinolone acetonide provided higher drug deposition in rabbit skin (epidermis and dermis) [Citation66]. Increased percutaneous delivery of progesterone, hydrocortisone in hairless mouse skin, comparable antimicrobial activity of butyl paraben was reported using liposomal formulations [Citation67,Citation68]. But liposomes were later shown to act merely as a skin drug “localizer” rather than as a drug “transporter” on topical application as they remain confined to the peripheral layer of SC instead deeply penetrating into the skin [Citation65,Citation69].
Transfersomes
To circumvent the problem of localization of liposomes on skin, new vesicular system was introduced to deliver drugs into deeper layers of skin. They are elastic or deformable in nature called ultradeformable liposomes or transfersomes [Citation70]. The membrane of transfersomes is constituted of phospholipid and edge activator, a single chain surfactant molecule. Sodium cholate, sodium deoxycholate, Span 60, Span 65, Span 80, Tween 20, Tween 60, Tween 80 and dipotassium glycyrrhizinate are employed as edge activators. Edge activator destabilizes the lipid bilayer of the vesicles and increases the deformability by lowering its interfacial tension [Citation71].
Several in vitro and in vivo studies have reported the ability of transfersomes to transport variety of drugs across the skin in therapeutic amounts [Citation72–74]. As compared to commercial hydrogel formulation, diclofenac entrapped within ultradeformable vesicles penetrated the skin in large amount, i.e. around 10 times more in concentration across the skin exhibiting its effect for prolonged period of time [Citation75]. Similarly, flexible vesicles were found superior for the transdermal transportation of cyclosporine A than conventional vesicles [Citation76]. As compared to plain drug solution, eight-fold increase in peak plasma concentration of levonorgestrel using optimized pro-transfersome gel (PTG) formulation was reported [Citation77]. With special reference to protein drugs, transfersomes were found to be equally effective in delivering them through the skin. In an study done using insulin loaded transfersomes, it was reported to show bioefficiency of at least 50% of the s.c. dose both in mice and humans and was able to achieve hypoglycaemia of minimum 30% of normal blood glucose level and approachable up to 50% [Citation78]. In another study, lecithin containing flexible insulin vesicles achieved hypoglycaemic effect by more than 50% for time period up to 18 h on mice skin [Citation76]. Similarly, encouraging results were reported in case of protein drug low molecular weight heparin (LMWH). Cationic LMWH elastic vesicles called flexosomes penetrated into deeper layers of the skin with three fold enhancement than control LMWH solution and was suggested as potential treatment therapy for superficial thrombosis, subcutaneous wounds, bruise and burns [Citation79]. The transport of Insulin and LMWH suggests transfersomes are sufficiently capable to deliver bio-macromolecular drugs across the skin, that to in therapeutic amount. Transfersomes were also studied for the delivery of entrapped antigens gap junction proteins (GJPs), protein in nature, to the immunologically active cells of the skin and lymph nodes. They were successful in inducing antigen-specific antibodies equivalent to subcutaneously injected vesicle suspensions which was also corroborated by the appreciable antibody titer formation after application of transfersomal tetanus toxoid formulations [Citation80].
Mechanism of penetration enhancement by transfersomes
The penetration of transfersomes may be explained by any one of the mechanism or combination of one or more of the mechanism mentioned in .
Figure 5. Transport mechanism of transfersomes: (i) movement of transfersomes through adsorption mechanism and (ii) transport of transfersomes using osmotic gradient mechanism.
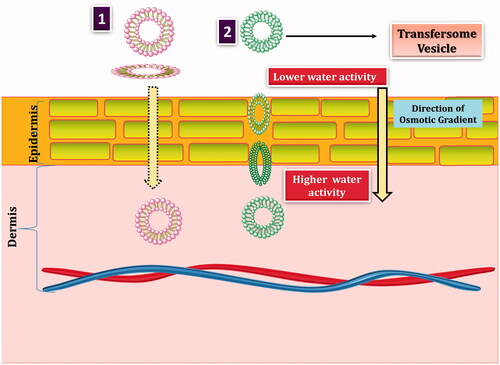
Transfersomes by virtue of its xerophobic nature, i.e. tendency to avoid dry surroundings enters into deeper layer of skin having higher moisture content than surface layer. The elasticity of membrane assists during the transport to breach narrow gap on the surface of skin and get transported intact. On the other hand, enhanced drug transfer by transfersomes can be attributed to its distribution on the skin surface following the adsorption on the skin [Citation81].
Microemulsions
Water in oil (W/O) microemulsions are thermodynamically stabilized dispersions composed of small (200 nm) water droplets dispersed within a continuous oil layer stabilized by the incorporation of a high concentration of surfactant/emulsifying molecules. Lipophilic surrounding in the external phase resembles the environment found in upper layer of skin, thus making it ideal candidate for application on to the skin surface [Citation82]. Additionally, the ease of administration of W/O micro-emulsions to the skin without the need for a delivery device makes it ideal for passive delivery of water-soluble molecules (proteins and peptides in this case) across the skin [Citation83]. Witting et al. studied the efficacy of topically applied TNF-receptor-Fc (TR-Fc) fusion protein, ALSTII, formulated within a W/O ME and found rapid penetration of the molecule into the skin immediately below the site of application, and rapid lateral movement to the distal area of the skin. The distribution studies revealed that up to 4 h there was very slow drainage of protein from dermal region into the circulation and other tissues of the body after which penetration into the circulation increased gradually giving a clear picture of penetration ability of microemulsions [Citation84]. Getie et al. in another study performed a comparative distribution study of desmopression acetate across the skin using W/O microemulsion and amphiphilic cream. The microemulsion successfully delivered about 6% of the applied dose whereas cream transported only 2% of the drug at a time period of 300 min. Besides this, the drug was localized on the peripheral surface of the skin and was present in higher amount on the skin treated with cream whereas the microemulsion was able to deliver drug to greater depth of the skin. This study not only validated the capability of microemulsions as a transdermal carrier system but also claimed its superiority over creams and gels in delivering drugs across the skin [Citation85]. In another study, the effect of subcutaneously and topically administered protein drugs (in the form of W/O microemulsion) insulin, IGF-I and GHRP-6, on body weight, muscle mass and weight of skin and fat was studied on obese mice of varying weight and it was observed that topically applied insulin, IGF-I and GHRP-6 all had an anabolic effect on the percentage muscle mass measured in treated animals when compared with control animals suggesting the passive transport of applied drugs across the skin. Had there been no transport, the observed effects would not have appeared [Citation83]. With all these illustrations, the role of W/O microemulsion in protein delivery cannot be denied but more understanding is the need of hour for its successful industrial application [Citation86,Citation87]. The superior transdermal delivery of microemulsions can be ascribed to two factors: high drug loading capacity of the microemulsions and penetration enhancing effect of constituents incorporated. Microemulsions have the unusual capacity of encompassing large amount of drugs, both hydrophilic and lipophilic in nature without a concurrent increase in vehicle affinity. This consequently leads to higher concentration gradient and higher transdermal flux from the microemulsions. On the other hand, the oil portion of surfactant applied, e.g. isopropyl palmitate (IPP) has generally been observed with augmenting permeation by disrupting the intercellular lipid structure of SC [Citation88,Citation89].
Nanoparticles
It is generally observed that the capability of nanoparticles and microparticles to penetrate across the skin is minimal. Human skin has been developed as a shield over million years of evolution to protect against natural nanoparticles like viruses, bacteria, dust, allergens, etc. In order to deliver drugs across the skin in the form of nanoparticles, this tough barrier needs to be breached [Citation90]. However, metal-based nanoparticles of less than 10 nm in size, including quantum dots and iron oxide nanoparticles are able to move across the skin. Huang et al. studied the effect of co-administered model protein drugs HRP (MW 45 kDa) and β-gal (MW 460 kDa) on the mice skin, in physical mixture with gold nanoparticles and demonstrated significant level of protein in epidermal layer with minute quantities in dermis after two hours of the application. Drugs entrapped within epidermis and dermis have the potential to further diffuse into deeper layer thereby suggesting it as a promising alternative to passive transdermal delivery. They further studied the potential of transcutaneous immunization on Balb/c mice by topical co-administering Au-NPs with ovalbumin antigen (OVA). It revealed that the topically applied OVA/Au-NPs physical mixture was highly immunogenic in mice, as anti-OVA IgG level was significantly increased (p>.05) over the course of the immunization schedule. Additionally, the level of IgG in animals receiving topical OVA/Au-NPs showed almost similar results in comparison with mice having standard vaccination by intramuscular injection [Citation91]. Additionally, neutral, anionic and cationic, spherical, and ellipsoid QDs (hydrodynamic radius 15–45 nm) dispersed in borate buffers (pH 8.7 and pH 9.0) when left in contact with porcine skin for 8 and 24 h showed that all spherical QDs penetrated the SC, and were found in the epidermis and dermis after 8 h as observed using confocal microscopy. Neutral and cationic ellipsoid QDs reached the VE in 8 h. In contrast, 24 h were needed to observe anionic ellipsoid QDs in the VE [Citation92]. Similarly, Chu et al. demonstrated ability of certain nanometersized CdTe QDs to permeate through mouse skin, and get accumulated in heart, liver, spleen, lung, kidney and brain under normal animal heat conditions. The concentration of the Cd atoms in these organs of mice coated with QDs retained a relatively high level for a long time (at least a week), suggesting slow rate of clearance of the QDs in vivo [Citation93]. Nanoparticles due to their distinct biological properties owing to its non-deformable and miniscule nature as compared to lipid or polymeric counter parts aid in transdermal transport. Metallic nanoparticles perturb the lipid phase on the membrane inducing gelled areas that enhances the lipid fluidity. The lipid fluidizing function of nanoparticles thus alters skin permeability and which in turn facilitates the passive transport to the deepest layers of the skin SC, the stratum granulosum and hair follicles and viable epidermis in exceptional cases [Citation91,Citation92,Citation94].
Lecithin organogels
Lecithin organogels (LOs) are clear, thermodynamically stable, thermo-reversible (sol-to-gel transition temperature as 40 °C), gels consisting of phospholipids (lecithin), appropriate organic solvent, and a polar solvent (mostly water) in suitable ratios. LOs, the jelly-like phases, consist of a three-dimensional network of entangled reverse cylindrical (polymer-like) micelles, which immobilizes the continuous or macroscopic external organic phase, thus turning a liquid into a gel [Citation95,Citation96]. Lecithin organogels are studied for their role as carriers of compounds both hydrophilic and hydrophobic in nature and evidenced its capability to transfer across the skin. Drugs like scopolamine, broxaterol, propranolol hydrochloride, fluoxetine hydrochloride, nicardipine, methimazole, piroxicam, anticancer agents and NSAIDs (diclofenac and indomethacin) are tested for transdermal delivery after incorporation into LOs and pluronic lecithin organogels (PLOs) and reported as an effective medium [Citation82,Citation95]. Besides being able to carry and deliver the drugs across the skin, it is imperative for the carrier system to retain structural and functional properties in case of proteins and peptides delivery. Lecithin organogels solubilize biologically active substances including large protein molecules while retaining their native structure and properties. For example, a significant amount of ascorbic acid or hydrophilic amino acids are solubilized in micelles of the lecithin gel without their deformation. In another study, immobilization of enzyme lipase in the lecithin/cyclohexane water organogel was done and studied for its catalytic activity against hydrolysis of triglyceride (tricaprylin) after incorporation. The catalytic activity of the enzyme was retained; however, the reaction occurred very slowly [Citation97]. LOs and PLOs have arguably been efficient delivery system for topical and transdermal application of low MW drugs. Their versatility for macromolecular drugs including protein and peptide medications shows a promising avenue ahead with existing evidences. Nevertheless, further studies need to be conducted to validate it as a useful transdermal vehicle for peptide and protein delivery. The enhanced flux of molecules is attributed to lecithin component of LOs and PLOs. Through low temperature SEM investigation and differential scanning calorimetry (DSC) studies, Dreher et al. showed intercellular lipid organization in SC was disturbed whereas corneocytes geometry remained unchanged [Citation89]. This disorganized lipid layer might thus provide requisite spaces for protein and peptide drugs to traverse across the skin.
Hyaluronic acid and its derivatives
Hyaluronic acid or hyaluronan, a natural linear polysaccharide of N-acetyl glucosamine and glucuronic acid in alternating sequence, has an average MW of approximately 2 × 105 to 107 Da [Citation98]. Human body comprises approximately 15 g of HA and human skin alone constitutes one third of it [Citation99]. When applied to the skin it provides beneficial effects such as skin hydration, elasticity regeneration and improved wound healing. Owing to its abundance and imparting positive benefits to the skin, HA and modified HA have been extensively investigated for their topical and transdermal applications. Solaraze®, i.e. 3% diclofenac in 2.5% HA gel was granted regulatory approval in the USA, Canada and Europe, for the topical treatment of actinic keratoses, which is the third most common skin complaint in the USA [Citation100].
Hyaluronic acid has been established as an effective topical delivery agent. The inclusion of HA in a topical formulation offers clear and unique potential in the delivery and localization of drugs to the skin [Citation100]. But their potential in transdermal delivery of drugs including biomacromolecules like proteins and peptides still remains an area diminutively explored. There are few literatures, however, where HA has been employed and studied for transdermal delivery of proteins. Yang et al. reported the receptor mediated transdermal delivery of human growth hormone (hGH), a peptide hormone, incorporating HA. They conjugated the hGH with aldehyde modified HA by specific coupling reaction and studied for its transdermal behaviour. The fluorescence microscopy successfully visualized the penetration of topically applied FITC labelled HA-hGH conjugate to the epidermis and even to the dermis in mice and pharmacokinetic studies reported the effective delivery of conjugate through the skin into the blood stream. On the other hand, FITC and hGH FITC conjugate could not penetrate the skin tissue mostly remaining on the surface of SC [Citation101]. Similarly, Martins et al. prepared solid in oil (s/o) nanodispersion of HA along with model protein BSA and reported ex vivo penetration in pig skin by fluorescence and confocal microscopy to the SC and even into the dermis layer [Citation102]. Witting et al. reported that HA acted as an enhancer for delivery of biomacromolecules in normal skin, usually mediated by a combination of co-transport, enhanced skin hydration and alterations in the SC properties. They studied the effect of HA hydrogels with varying MWs (5 kDa, 100 kDa, 1 MDa) on the skin absorption of the model protein bovine serum albumin (BSA). Five kilodaltons HA led to a higher penetration enhancement of BSA into the viable epidermis (with an enhancement factor of 7.5) compared to 100 kDa and 1 MDa. Formulations containing low MW HA (5 kDa HA) altered the SC barrier properties through keratin structural changes (α-helix to β-sheet interconversion of keratin in the SC) and augmented the skin hydration, in particular at higher concentrations (5% and 10% HA) . This showed the ability of low MW HA to breach the disrupted SC while high MW HA could not do so. Also, close contact was observed between HA and BSA molecules both in the SC and in the epidermis suggesting co-transport by low MW HA as the most credible explanation [Citation98]. HA modified transfersomes were prepared by Hou and Kong by assembling amphiphilic HA derivative on the surface of a transfersome to form multilayered, spherical and highly flexible HA-T. It significantly increased the transdermal penetration of doxorubicin in rat skin and lead to higher accumulation in lymphatics in vivo, such as spleen and lymph nodes [Citation46] (). Hyaluronic acid has shown the versatility in improving transdermal delivery of macromolecules by different mechanisms as described above. They can be conjugated with protein drugs, engineered on the surface of transfersomes, assembled in the form of s/o nano-dispersions, formulated in the form of hydrogels for circumventing the SC. This adaptability of HA is promising and can be anticipated for its greater position in transdermal delivery in near future.
Figure 6. Schematic illustration of hyaluronic acid modified transfersomes for drug delivery towards lymphatics through the transdermal route.
Mechanism of penetration
The transport mechanism of HA in the transdermal transport of protein drugs has no any definitive explanations yet. Nonetheless, there are plausible suggestions to co-relate the observed effect of enhanced permeation. First, HA being extremely hydrophilic in nature hydrates the skin to the extent that it loosens the cluster of densely packed corneocytes opening up intercellular spaces as well as intracellular spaces promoting the passage of molecules through them [Citation103]. On the other hand, major HA receptors distributed on the skin (CD44 and hyaladherin on fibroblasts and keratinocytes) mediate the co-transport of HA derivatives inside the dermal layer [Citation104]. Hydrophobic portion of the HA chain might have role in the transport process of HA and its derivatives through intercellular lipid region due to its close affinity with the lipid layer [Citation101].
Miscellaneous approaches
Prodrug
Prodrug involves the transient or reversible modification of the physicochemical properties of compounds through chemical derivatization to augment the solubility or bioavailability profile or to provide more stability compared to the parent drug while preserving the pharmacological properties of the parent drug. There are reports suggesting the derivatization of the bioactive peptides to produce prodrugs that possess enhanced physicochemical properties compared to original compounds with regard to delivery and metabolic stability. A prerequisite for success of the prodrug is the reliable conversion of prodrug to the parent drug through either enzymatic or non-enzymatic catalysed reaction, once the barrier to delivery has been circumvented. To preserve the physical and chemical integrity remains additional responsibility during prodrug fabrication for proteins [Citation105,Citation106]. There is plethora of examples where pro-drugs have been synthesized of proteins and have been studied for the stability and chemical reversibility at plasma after application. The pyroglutamyl peptide and its derivatives for the prodrug approach to protect a peptide against specific enzymatic cleavage by pyroglutamyl aminopeptidase and 4-imiadazolidinones derivatives formed by condensing compounds containing an alpha-amino amide moiety, abundant in most peptide groups with aldehydes or ketones, are few of them. There is already one 4-imidazolidinone prodrug derivative in clinical use, namely hetacillin, formed by condensation of ampicillin with acetone [Citation107]. In this review, however, we have focused on the increased flux of peptides and proteins prodrugs across membranes rather than on solubility enhancement or pharmacokinetic superiority of them. Bundgaard has successfully transported protein TRH across human skin. Using the lipophilic prodrug of TRH, i.e. N-octyloxycarbonyl-TRH, they achieved a very efficient delivery of TRH through human skin. They applied prodrug derivative in an aqueous solution (pH 6.0) at a concentration of 5% and reported a steady state flux of 0.045 µmol h−1 cm−2 corresponding to 16 µgm h−1 cm−2. Essentially, all of the prodrug applied was converted to the parent TRH during diffusion through the human skin samples. Transdermal patch of area 20 cm2 having a flux of 16 µgm h−1 cm−2 is showing to deliver 320 µg TRH/h or 7.6 mg TRH over 24 h which is in equivalent to the amount that is often given by infusion or injection of TRH during 24 h. The good skin penetration behaviour observed for the N-octyloxycarbonyl prodrug derivative was attributed most likely to its combination of high water solubility (>10%) and lipophilicity [Citation105]. Proteins also cross biological membranes by carrier-mediated processes [Citation108]. Conjugation of proteins or peptides by such carriers which selectively transport them across the biological membranes might be useful in delivery across the skin, ricin transport being an example. The toxic protein, ricin, is transported across the cell membrane via binding to the ricin B chain found on the surface followed by internalization. Upon entering the cell, the active component, the ricin A chain, is liberated where it exerts its toxicological effects. Therefore, ricin behaves essentially as a prodrug of ricin A chain. The use of monoclonal antibody–ricin A chain conjugates for selective delivery of this peptide toxin has also been studied. Similar scenarios can be envisaged where other prodrugs of polypeptides, including proteins, could be delivered conjugating it with membrane transporting carrier involving similar carrier-mediated mechanisms [Citation106].
Skin penetration modifiers
Numerous researches analyzing transcutol as a skin penetration modifier depicted that DEGEE increased the solubility of permeant in the skin without remarkably affecting its diffusivity in the skin, i.e. SC. For the permeants dexamethasone and hydrocortisone, the presence of DEGEE resulted in enhanced skin retention although the permeability and therefore the systemic uptake were significantly decreased. This effect has been called the intracutaneous depot and can be conceptualized as DEGEE increasing the reservoir capacity of the SC. Thus, although DEGEE is a skin penetration modifier, it is not accurate to describe DEGEE as a skin penetration enhancer. Transcutol mainly solubilizes the drug molecules through co-solvent effect and increases the cutaneous retention of drug molecules. The combined effect of both of the above thus helps to enhance drug permeation. Few solvents like ethanol and propylene glycol are penetration enhancers which when employed at adequate concentrations enhance the penetration rate of dissolved drugs. Although enhancement ranges from minimal to dramatic, use of these solvents universally enhances skin penetration. In contrast, DEGEE may increase penetration or it may significantly decrease systemic uptake of a dissolved drug. The exact mechanism by which DEGEE will modify the penetration of drugs via skin can not be anticipated [Citation109,Citation110].
Choice of active or passive delivery
The choice of active versus passive method for the transdermal delivery including that of protein and peptide drugs involves considerable ambiguity. Active delivery methods augment the transport process by either providing additional force for transportation or generating permanent sites through mechanical disruption of skin for smooth passage of drugs. By doing so, they reach the systemic circulation in shorter period of time compared to passive method of delivery. Additionally, these technologies available can be manoeuvred to meet the patient specific demands. However, they require sophistication in terms of design, functioning and control. Aspects like incorporation of electricity, temperature or other energy medium along with sensor mechanism for control of delivery make active method implementation a highly challenging task. Contrary to this, passive method of delivery is more patient friendly. Passive approaches like CPEs, nanocarriers or peptides are relatively easy to apply and cause minimal dermal damage. They can be fabricated to apply in larger surface areas when necessitated to deliver higher amount of drugs. In addition, they do not require strenuous effort in terms of development and implementation unlike active ones. But, passive method after application requires certain time to reach the systemic circulation. This delayed onset of action, sometimes up to several hours, after application is critical in cases where patients demand immediate care like insulin therapy in diabetic patients [Citation111].
In such scenario, approaches incorporating the benefits of both active and passive methods of delivery rather than sticking to one particular type seem to be the wise alternative. By combining, liposomes with microneedles and iontophoresis, Bashyal and Lee showed that the flux of insulin was enhanced by 713.3-fold higher as compared to passive permeation [Citation112]. Wang et al. reviewed the combination of iontophoresis with multiple other techniques like chemical enhancers, even with other active strategies like sonophoresis, microneedle, electroporation and suggested this combination of techniques helps in more compliant and accurate delivery of macromolecules [Citation113]. Use of microneedles along with iontophoresis, CPEs with ultrasound, low frequency ultrasound with iontophoresis are some of the examples where combination approach was used for relatively better transdermal transport [Citation114–116]. Le et al. further emphasized the fact that the use of combination strategies culminates in consumption of less energy for active method which otherwise demanded higher amount of energy when used alone. It also provides the opportunity for better control during therapy as in combination of ultrasound and iontophoresis, where ultrasound regulates the permeability of skin whereas iontophoresis control the flux [Citation117].
Conclusions and future perspectives
Delivery of protein and peptides through transdermal route is a herculean task as they pose number of hurdles owing to unfavourable physicochemical properties (high MW and hydrophilicity), instability, complex structural orientation and potency. To circumvent key barrier of skin, i.e. SC for delivery of transdermal therapeutics, some mechanical methods like iontophoresis, thermal ablation, jet injectors, etc. have been explored but their complex design and uneasy application limited their use and further development. Simpler techniques were sought after that could possibly breach the barrier properties of skin as well as involve negligible risks of irreversible damage to the skin. Methods using peptide derivatives, prodrugs, HA derivatives and other techniques reviewed above have shown promising results in this area. These techniques clearly are not in a phase to reap their benefits but definitely have provided a new horizon to bring forth newer developments for better delivery approaches across skin. Chemical enhancers, nanocarriers, certain biological peptides and miscellaneous approaches including prodrugs and LOs are under extensive exploration for non-invasive transdermal delivery. Moreover, meticulous delivery of therapeutic peptide and proteins through the skin is to be warranted for uncompromised safety and efficacy. Scientists are in a quest to revisit such undiscovered areas which could pave newer dimensions for non-invasive delivery across the skin.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–672.
- Walter JR, Xu S. Therapeutic transdermal drug innovation from 2000 to 2014: current status and outlook. Drug Discov Today. 2015;20:1293–1299.
- Watkinson AC, Kearney M-C, Quinn HL, et al. Future of the transdermal drug delivery market – have we barely touched the surface? Expert Opin Drug Deliv. 2016;13:523–532.
- Wiedersberg S, Guy RH. Transdermal drug delivery: 30+ years of war and still fighting! J Control Release. 2014;190:150–156.
- Kinch MS. An overview of FDA-approved biologics medicines. Drug Discov Today. 2015;20:393–398.
- Katikaneni S. Transdermal delivery of biopharmaceuticals: dream or reality? Ther Deliv. 2015;6:1109–1116.
- Kalluri H, Banga AK. Transdermal delivery of proteins. AAPS PharmSciTech. 2011;12:431–441.
- Munch S, Wohlrab J, Neubert RHH. Dermal and transdermal delivery of pharmaceutically relevant macromolecules. Eur J Pharm Biopharm. 2017;119:235–242.
- Wong TW, Nor Khaizan A. Physicochemical modulation of skin barrier by microwave for transdermal drug delivery. Pharm Res. 2013;30:90–103.
- Herwadkar A, Banga A. Transdermal delivery of peptides and proteins. In: Van Der Walle C, editor. Peptide and protein delivery. Chap. 4. Amsterdam: Elsevier; 2011. p. 69–86.
- Perumal O, Murthy S, Kalia Y. Turning theory into practice: the development of modern transdermal drug delivery systems and future trends. Skin Pharmacol Physiol. 2013;26:331–342.
- Bouwstra JA, Dubbelaar FE, Gooris GS, et al. The lipid organisation in the skin barrier. Acta Derm Venereol Suppl (Stockh). 2000;208:23–30.
- Bouwstra JA, Honeywell-Nguyen PL, Gooris GS, et al. Structure of the skin barrier and its modulation by vesicular formulations. Prog Lipid Res. 2003;42:1–36.
- Bouwstra JA, Honeywell-Nguyen PL. Skin structure and mode of action of vesicles. Adv Drug Deliv Rev. 2002;54(Suppl 1):S41–S55.
- Boncheva M. The physical chemistry of the stratum corneum lipids. Int J Cosmet Sci. 2014;36:505–515.
- Matsui T, Amagai M. Dissecting the formation, structure and barrier function of the stratum corneum. Int Immunol. 2015;27:269–280.
- van Smeden J, Janssens M, Gooris GS, et al. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim Biophys Acta. 2014;1841:295–313.
- Madison KC. Barrier function of the skin: “la raison d'être” of the epidermis. J Invest Dermatol. 2003;121:231–241.
- Barry BW. Action of skin penetration enhancers—the lipid protein partitioning theory. Int J Cosmet Sci. 1988;10:281–293.
- Williams AC, Barry BW. Penetration enhancers. Adv Drug Deliv Rev. 2012;64:128–137.
- Dragicevic N, Atkinson JP, Maibach HI. Chemical penetration enhancers: classification and mode of action. In: Dragicevic N, Maibach H, editors. Percutaneous penetration enhancers chemical methods in penetration enhancement. Berlin, Heidelberg: Springer; 2015. p. 11–27.
- Pathan IB, Setty CM. Chemical penetration enhancers for transdermal drug delivery systems. Trop J Pharm Res. 2009;8(2):173–179.
- Klimentová J, Hrabálek A, Vávrová K, et al. Synthesis and transdermal penetration-enhancing activity of carbonic and carbamic acid esters—comparison with transkarbam 12. Bioorg Med Chem Lett. 2006;16:1981–1984.
- Parhi R, Suresh P, Mondal S, et al. Novel penetration enhancers for skin applications: a review. CDD. 2012;9:219–230.
- Bhatia KS, Singh J. Mechanism of transport enhancement of LHRH through porcine epidermis by terpenes and iontophoresis: permeability and lipid extraction studies. Pharm Res. 1998; 15:1857–1862.
- Smyth HD, Becket G, Mehta S. Effect of permeation enhancer pretreatment on the iontophoresis of luteinizing hormone releasing hormone (LHRH) through human epidermal membrane (HEM). J Pharm Sci. 2002;91:1296–1307.
- Moghadam SH, Saliaj E, Wettig SD, et al. Effect of chemical permeation enhancers on stratum corneum barrier lipid organizational structure and interferon alpha permeability. Mol Pharm. 2013;10:2248–2260.
- Bhatia KS, Gao S, Singh J. Effect of penetration enhancers and iontophoresis on the FT-IR spectroscopy and LHRH permeability through porcine skin. J Control Release. 1997;47:81–89.
- Kumar S, Zakrewsky M, Chen M, et al. Peptides as skin penetration enhancers: mechanisms of action. J Control Release. 2015;199:168–178.
- Namjoshi S, Benson HA. Cyclic peptides as potential therapeutic agents for skin disorders. Biopolymers. 2010;94:673–680.
- Menegatti S, Zakrewsky M, Kumar S, et al. De novo design of skin-penetrating peptides for enhanced transdermal delivery of peptide drugs. Adv Healthcare Mater. 2016;5:602–609.
- Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206.
- Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529–545.
- Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–393.
- Chugh A, Eudes F, Shim YS. Cell-penetrating peptides: nanocarrier for macromolecule delivery in living cells. IUBMB Life. 2010;62:183–193.
- Dupont E, Prochiantz A, Joliot A. Penetratin story: an overview. Methods Mol Biol. 2011;683:21–29.
- Lindgren M, Langel U. Classes and prediction of cell-penetrating peptides. Methods Mol Biol. 2011;683:3–19.
- Elmquist A, Hansen M, Langel U. Structure–activity relationship study of the cell-penetrating peptide pVEC. Biochim Biophys Acta. 2006;1758:721–729.
- Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840.
- Morris MC, Deshayes S, Heitz F, et al. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100:201–217.
- Pooga M, Hallbrink M, Zorko M, et al. Cell penetration by transportan. FASEB J. 1998;12:67–77.
- Rousselle C, Smirnova M, Clair P, et al. Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: saturation kinetics and specificity. J Pharmacol Exp Ther. 2001;296:124–131.
- Jain A, Shah SG, Chugh A. Cell penetrating peptides as efficient nanocarriers for delivery of antifungal compound, natamycin for the treatment of fungal keratitis. Pharm Res. 2015;32:1920–1930.
- Cohen-Avrahami M, Shames AI, Ottaviani MF, et al. HIV-TAT enhances the transdermal delivery of NSAID drugs from liquid crystalline mesophases. J Phys Chem B. 2014;118:6277–6287.
- Jin LH, Bahn JH, Eum WS, et al. Transduction of human catalase mediated by an HIV-1 TAT protein basic domain and arginine-rich peptides into mammalian cells. Free Radic Biol Med. 2001;31:1509–1519.
- Hou L, Kong M. Enhanced transdermal lymphatic drug delivery of hyaluronic acid modified transfersome for tumor metastasis therapy. J Control Release. 2015;213:e77.
- Nasrollahi SA, Taghibiglou C, Azizi E, et al. Cell-penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des. 2012;80:639–646.
- Gennari CG, FranzeÌ S, Pellegrino S, et al. Skin penetrating peptide as a tool to enhance the permeation of heparin through human epidermis. Biomacromolecules. 2015;17:46–55.
- de Cogan F, Hill LJ, Lynch A, et al. Topical delivery of anti-VEGF drugs to the ocular posterior segment using cell-penetrating peptides CPP delivery of anti-VEGF drugs to the retina. Invest Ophthalmol Vis Sci. 2017;58:2578–2590.
- Shi N-Q, Qi X-R, Xiang B, et al. A survey on “Trojan Horse” peptides: opportunities, issues and controlled entry to “Troy”. J Control Release. 2014;194:53–70.
- Desai P, Patlolla RR, Singh M. Interaction of nanoparticles and cell-penetrating peptides with skin for transdermal drug delivery. Mol Membr Biol. 2010;27:247–259.
- Richard JP, Melikov K, Vives E, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590.
- Di Pisa M, Chassaing G, Swiecicki J-M. Translocation mechanism(s) of cell-penetrating peptides: biophysical studies using artificial membrane bilayers. Biochemistry. 2014;54:194–207.
- Kumar S, Narishetty ST, Tummala H. Peptides as skin penetration enhancers for low molecular weight drugs and macromolecules. In: Dragicevic N, Maibach H, editors. Percutaneous penetration enhancers chemical methods in penetration enhancement. Berlin, Heidelberg: Springer; 2015. p. 337–352.
- Hsu T, Mitragotri S. Delivery of siRNA and other macromolecules into skin and cells using a peptide enhancer. Proc Natl Acad Sci. 2011;108:15816–15821.
- Chen M, Gupta V, Anselmo AC, et al. Topical delivery of hyaluronic acid into skin using SPACE-peptide carriers. J Control Release. 2014;173:67–74.
- Chen M, Kumar S, Anselmo AC, et al. Topical delivery of cyclosporine A into the skin using SPACE-peptide. J Control Release. 2015;199:190–197.
- Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA. 1987;84:5449–5453.
- Kim YC, Ludovice PJ, Prausnitz MR. Transdermal delivery enhanced by magainin pore-forming peptide. J Control Release. 2007;122:375–383.
- Kim Y-C, Ludovice PJ, Prausnitz MR. Optimization of transdermal delivery using magainin pore-forming peptide. J Phys Chem Solids. 2008;69:1560–1563.
- Kim YC, Late S, Banga AK, et al. Biochemical enhancement of transdermal delivery with magainin peptide: modification of electrostatic interactions by changing pH. Int J Pharm. 2008;362:20–28.
- Chen Y, Shen Y, Guo X, et al. Transdermal protein delivery by a coadministered peptide identified via phage display. Nat Biotechnol. 2006;24:455–460.
- Lopes LB, Carvalho V, de Lemos DP. Potential of peptide-based enhancers for transdermal delivery. CPD. 2015;21:2814–2822.
- Kristensen M, Birch D, Mørck Nielsen H. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. IJMS. 2016;17:185.
- Touitou E. Drug delivery across the skin. Expert Opin Biol Ther. 2002;2:723–733.
- Mezei M, Gulasekharam V. Liposomes—a selective drug delivery system for the topical route of administration: lotion dosage form. Life Sci. 1980;26:1473–1477.
- Ganesan MG, Weiner ND, Flynn GL, et al. Influence of liposomal drug entrapment on percutaneous absorption. Int J Pharm. 1984;20:139–154.
- Konno T. Physical and chemical changes of medicinals in mixtures with adsorbents in the solid state. IV: study on reduced-pressure mixing for practical use of amorphous mixtures of flufenamic acid. Chem Pharm Bull. 1990;38:2003–2007.
- Honeywell-Nguyen PL, Bouwstra JA. Vesicles as a tool for transdermal and dermal delivery. Drug Discov Today Technol. 2005;2:67–74.
- Cevc G, Blume G. Lipid vesicles penetrate into intact skin owing to the transdermal osmotic gradients and hydration force. Biochim Biophys Acta (BBA)-Biomembr. 1992;1104:226–232.
- Elsayed MM, Abdallah OY, Naggar VF, et al. Lipid vesicles for skin delivery of drugs: reviewing three decades of research. Int J Pharm. 2007;332:1–16.
- Ahad A, Al-Saleh AA, Al-Mohizea AM, et al. Formulation and characterization of Phospholipon® 90 G and Tween® 80 based transfersomes for transdermal delivery of eprosartan mesylate. Pharm Dev Technol. Forthcoming. [27 p.]. DOI: https://doi.org/10.1080/10837450.2017.1330345.
- Abdellatif AA, Tawfeek HM. Transfersomal nanoparticles for enhanced transdermal delivery of clindamycin. AAPS PharmSciTech. 2016;17:1067–1074.
- Shreya AB, Managuli RS, Menon J, et al. Nano-transfersomal formulations for transdermal delivery of asenapine maleate: in vitro and in vivo performance evaluations. J Liposome Res. 2016; 26:221–232.
- Cevc G, Blume G. New, highly efficient formulation of diclofenac for the topical, transdermal administration in ultradeformable drug carriers, transfersomes. Biochim Biophys Acta. 2001; 1514:191–205.
- Guo J, Ping Q, Sun G, et al. Lecithin vesicular carriers for transdermal delivery of cyclosporin A. Int J Pharm. 2000;194:201–207.
- Jain S, Sapre R, Tiwary AK, et al. Proultraflexible lipid vesicles for effective transdermal delivery of levonorgestrel: development, characterization, and performance evaluation. AAPS PharmSciTech. 2005;6:E513–E522.
- Cevc G, Gebauer D, Stieber J, et al. Ultraflexible vesicles, transfersomes, have an extremely low pore penetration resistance and transport therapeutic amounts of insulin across the intact mammalian skin. Biochim Biophys Acta (BBA)-Biomembr. 1998; 1368:201–215.
- Song YK, Kim CK. Topical delivery of low-molecular-weight heparin with surface-charged flexible liposomes. Biomaterials. 2006;27:271–280.
- Paul A, Cevc G, Bachhawat BK. Transdermal immunization with large proteins by means of ultradeformable drug carriers. Eur J Immunol. 1995;25:3521–3524.
- Cevc G, Chopra A. Deformable (Transfersome®) vesicles for improved drug delivery into and through the skin. In: Dragicevic N, Maibach H, editors. Percutaneous penetration enhancers chemical methods in penetration enhancement. Berlin, Heidelberg: Springer; 2016. p. 39–59.
- Murdan S. Organogels in drug delivery. Expert Opin Drug Deliv. 2005;2:489–505.
- Russell-Jones G, Himes R. Water-in-oil microemulsions for effective transdermal delivery of proteins. Expert Opin Drug Deliv. 2011;8:537–546.
- Witting M, Obst K, Friess W, et al. Recent advances in topical delivery of proteins and peptides mediated by soft matter nanocarriers. Biotechnol Adv. 2015;33:1355–1369.
- Getie M, Wohlrab J, Neubert RH. Dermal delivery of desmopressin acetate using colloidal carrier systems. J Pharm Pharmacol. 2005;57:423–427.
- Himes R, Lee S, McMenigall K, et al. Reduction in inflammation in the footpad of carrageenan treated mice following the topical administration of anti-TNF molecules formulated in a micro-emulsion. J Control Release. 2010;145:210–213.
- Goebel A, Schmaus G, Neubert R, et al. Dermal peptide delivery using enhancer molecules and colloidal carrier systems—Part I: carnosine. Skin Pharmacol Physiol. 2012;25:281–287.
- Kreilgaard M. Influence of microemulsions on cutaneous drug delivery. Adv Drug Deliv Rev. 2002;54(Suppl 1):S77–S98.
- Dreher F, Walde P, Walther P, et al. Interaction of a lecithin microemulsion gel with human stratum corneum and its effect on transdermal transport. J Control Release. 1997;45:131–140.
- Prow TW, Grice JE, Lin LL, et al. Nanoparticles and microparticles for skin drug delivery. Adv Drug Deliv Rev. 2011;63:470–491.
- Huang Y, Yu F, Park YS, et al. Co-administration of protein drugs with gold nanoparticles to enable percutaneous delivery. Biomaterials. 2010;31:9086–9091.
- Baroli B, Ennas MG, Loffredo F, et al. Penetration of metallic nanoparticles in human full-thickness skin. J Invest Dermatol. 2007;127:1701–1712.
- Chu M, Wu Q, Wang J, et al. In vitro and in vivo transdermal delivery capacity of quantum dots through mouse skin. Nanotechnology. 2007;18:455103.
- Verma A, Jain A, Hurkat P, et al. Transfollicular drug delivery: current perspectives. Res Rep Transderm Drug Deliv. 2016;5:1–17.
- Kumar R, Katare OP. Lecithin organogels as a potential phospholipid-structured system for topical drug delivery: a review. AAPS PharmSciTech. 2005;6:E298–E310.
- Almeida H, Amaral MH, Lobão P, et al. Pluronic® F-127 and Pluronic Lecithin Organogel (PLO): main features and their applications in topical and transdermal administration of drugs. J Pharm Pharm Sci. 2012;15:592–605.
- Murashova N, Yurtov E. Lecithin organogels as prospective functional nanomaterial. Nanotechnol Russia. 2015;10:511–522.
- Witting M, Boreham A, Brodwolf R, et al. Interactions of hyaluronic acid with the skin and implications for the dermal delivery of biomacromolecules. Mol Pharm. 2015;12:1391–1401.
- Essendoubi M, Gobinet C, Reynaud R, et al. Human skin penetration of hyaluronic acid of different molecular weights as probed by Raman spectroscopy. Skin Res Technol. 2016;22:55–62.
- Brown MB, Jones SA. Hyaluronic acid: a unique topical vehicle for the localized delivery of drugs to the skin. J Eur Acad Dermatol Venerol. 2005;19:308–318.
- Yang JA, Kim ES, Kwon JH, et al. Transdermal delivery of hyaluronic acid – human growth hormone conjugate. Biomaterials. 2012;33:5947–5954.
- Martins M, Azoia NG, Shimanovich U, et al. Design of novel BSA/hyaluronic acid nanodispersions for transdermal pharma purposes. Mol Pharm. 2014;11:1479–1488.
- Dragicevic N, Maibach HI. Modification of the stratum corneum. In: Percutaneous penetration enhancers chemical methods in penetration enhancement. Berlin, Heidelberg: Springer; 2015.
- Bourguignon LY, Singleton PA, Diedrich F. Hyaluronan-CD44 interaction with Rac1-dependent protein kinase N-γ promotes phospholipase Cγ1 activation, Ca2+ signaling, and cortactin-cytoskeleton function leading to keratinocyte adhesion and differentiation. J Biol Chem. 2004;279:29654–29669.
- Bundgaard H. (C) Means to enhance penetration: (1) prodrugs as a means to improve the delivery of peptide drugs. Adv Drug Deliv Rev. 1992;8:1–38.
- Oliyai R, Stella VJ. Prodrugs of peptides and proteins for improved formulation and delivery. Annu Rev Pharmacol Toxicol. 1993;33:521–544.
- Jusko WJ, Lewis GP. Comparison of ampicillin and hetacillin pharmacokinetics in man. J Pharm Sci. 1973;62:69–76.
- van Deurs B, Hansen SH, Olsnes S, et al. Protein uptake and cytoplasmic access in animal cells. In: Audus KL, Raub TJ, editors. Biological barriers to protein delivery. Boston (MA): Springer US; 1993. p. 71–104.
- Osborne DW. Diethylene glycol monoethyl ether: an emerging solvent in topical dermatology products. J Cosmet Dermatol. 2011;10:324–329.
- Mura P, Faucci M, Bramanti G, et al. Evaluation of transcutol as a clonazepam transdermal permeation enhancer from hydrophilic gel formulations. Eur J Pharm Sci. 2000;9:365–372.
- Arora A, Prausnitz MR, Mitragotri S. Micro-scale devices for transdermal drug delivery. Int J Pharm. 2008;364:227–236.
- Bashyal S, Lee S. Delivery of biopharmaceuticals using combination of liposome and iontophoresis: a review. J Pharm Investig. 2015;45:611–624.
- Wang Y, Thakur R, Fan Q, et al. Transdermal iontophoresis: combination strategies to improve transdermal iontophoretic drug delivery. Eur J Pharm Biopharm. 2005;60:179–191.
- Vemulapalli V, Yang Y, Friden PM, et al. Synergistic effect of iontophoresis and soluble microneedles for transdermal delivery of methotrexate. J Pharm Pharmacol. 2008;60:27–33.
- Johnson ME, Mitragotri S, Patel A, et al. Synergistic effects of chemical enhancers and therapeutic ultrasound on transdermal drug delivery. J Pharm Sci. 1996;85:670–679.
- Jain A, Tiwari A, Verma A, et al. Ultrasound-based triggered drug delivery to tumors. Drug Deliv Transl Res. 2018;8:150–164.
- Le L, Kost J, Mitragotri S. Combined effect of low-frequency ultrasound and iontophoresis: applications for transdermal heparin delivery. Pharm Res. 2000;17:1151–1154.