
Abstract
In this study, we demonstrate cell culture platforms that can provide a microenvironment similar to in vivo conditions so that in vivo-compatible drug testing results can be obtained from the in vitro experiments. To realize such in vivo microenvironment-mimetic platforms, different culture platforms such as a three-dimensional (3D) cell aggregate film, fluidic environment within a microfluidic system or extracellular matrix (ECM) coating were established. The tumor cell growth rate and sensitivity to doxorubicin (DOX) were studied using the glioblastoma cell line T98G. When 3 D spheroids were cultured, they grew significantly slower than under other culture conditions. When the cells were treated with DOX, the anticancer drug could not efficiently penetrate the 3 D spheroids to inhibit cell growth. When cultured on the Matrigel-coated culture vessel, T98G cells grew even in the presence of DOX, demonstrating chemoresistance. Nonetheless, in the 2D culture plate and in the microfluidic chip, cell growth decreased with DOX treatment and the binding ability was lost. These results indicate that the cells reacted differently to the same anticancer drug depending on the culture microenvironment. We believe that the development of a more physiologically relevant tumor cell culture platform will lead to more reliable antitumor drug responses.
Introduction
The drug discovery pipeline in the pharmaceutical industry requires substantial resources and manpower because it is time consuming and costly [Citation1,Citation2]. Generally, it is known that developing a new drug requires approximately 12–15 years of time and costs over $100 million before the drug can be utilized in the clinical stage [Citation3]. During the development of such new drugs, numerous drug candidates (usually a few thousand) are screened in a high-throughput manner using cells cultured in plastic culture dishes and fewer than 100 effective candidates are selected. Then, the cytotoxicity of drug candidates is tested in a dose-dependent manner using the same two-dimensional (2D) culture dishes [Citation4,Citation5]. In the next preclinical stage, ∼115 million animals are used worldwide each year for experiments to ensure these drugs safety and efficacy [Citation6]. In spite of these long and costly processes, many drugs that have passed the in vitro and animal studies have ultimately failed in the clinical trials [Citation7,Citation8]. This situation indicates that the results obtained in vitro and in animal studies are inconsistent with the drug responses in humans [Citation9,Citation10].
According to the US Food and Drug Administration (FDA), only 8% of the drugs that have passed animal testing have had their safety and effectiveness proven [Citation11]. Among the pharmaceutical classes of compounds that repeatedly fail as described above, anticancer drugs are the most common [Citation12,Citation13]. The main reason for this high failure rate is that the existing in vitro models do not precisely recapitulate the complex microenvironment of a human body, which operates under 3 D, extracellular-rich and fluidic conditions. Furthermore, animal models have inherent limitations such as genetic heterogeneity with humans [Citation14,Citation15]. Therefore, to obtain in vivo-compatible results at the preclinical stage that can be translated to the actual clinical trials, a new platform is urgently needed. With in vivo condition-mimicking platforms, the existing problems can be overcome and the cost and time required for drug development can be reduced.
In this study, we selected a brain tumor cell line with the aim to develop a platform that simulates the microenvironment of native tumor cells in vitro. There are several distinctive features in the native tumor microenvironment [Citation16,Citation17]. First, in general, tumor cells do not grow in a 2D morphology; rather, they grow as 3D cell aggregates [Citation18,Citation19]. Second, the tumor mass is consistently exposed to the fluidic shear stress caused by interstitial flow [Citation20]. It is noted that fluidic shear stress is an important factor in the growth and metastasis of tumors and tumor cells are sensitive to shear stress [Citation21]. Third, tumor cells interact with the surrounding extracellular matrix (ECM) and stromal cells using receptor or junctional proteins [Citation22,Citation23]. These cell-ECM and cell-cell interactions are important because not only can they modulate the growth kinetics of tumor cells by the limited diffusion of oxygen and nutrients in the tumor, but they can also induce cell adhesion-mediated drug resistance [Citation24] and subsequently change the sensitivity upon drug treatment [Citation25,Citation26]. Therefore, we attempted to implement these native features of the tumor microenvironment into the engineered tumor cell culture platforms. For this purpose, T98G, a brain tumor (glioblastoma) cell line, was cultured on each platform including the 3D tumor spheroid film, the microfluidic chambers and the ECM-coated culture vessel and the sensitivity to (and specificity of) DOX was evaluated in each environment.
Material and methods
Cancer cell preparation
Human brain tumor cell line T98G was purchased from Korean Cell Line Bank (Seoul, Korea). The tumor cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% of fetal bovine serum and 1% of a penicillin solution in an incubator set to 5% carbon di oxide (CO2) and 37 °C. To transfer the cells into engineered cell culture platforms, the cultured cells were removed from the culture dish using a 0.25% trypsin-EDTA solution (Life Technologies, Grand Island, NY) and the cell density was adjusted to a suitable value for each cell culture platform.
3D spheroid cancer cell culture using the 3D SpheroFilm™
A commercially available SpheroFilm™ (Incyto, Republic of Korea) was purchased and used to grow cancer cells in a 3D spheroid form. SpheroFilm™ is made of a silicone elastomer and has an array of concave microwells that helps grow cells into large 3D spheroid structures. When cells are dispensed above the microwell array, they gravitate into the microwells. If the cell–cell affinity is stronger than that between cells and the surface of the film, the cells spontaneously aggregate into a spheroid form. The microwells have an internal diameter of 500 μm and a depth of 100 μm. To control cell density within the microwells, the films were cut into 0.5 cm2 pieces and placed in 96-well plates. Prior to cell culture, 100% ethanol was introduced to avoid the formation of air bubbles. The ethanol was removed after 10 min and the wells were rinsed thoroughly with phosphate-buffered saline (PBS). After the washing step, the cells were seeded uniformly on the SpheroFilm™ with a cell density of 106 cells/mL and the cell-seeded film was immersed in the culture medium. The system was kept in an incubator for 24 h. The culture medium was changed every day in such a manner that one-third of the culture medium contained in the existing microwells was aspirated and the same volume was replenished.
Live-dead cell staining
To directly visualize the viability of 3D cancer spheroids, we stained the cells with two dyes. The Hoechst nuclear stain was applied to identify the total number of cells and the dead cells were identified by propidium iodide (PI) staining. First, cells were seeded on SpheroFilm™, and spheroid cells were collected every day from day 1 to 4 and transferred onto a glass slide. A drop of NucBlue™ Live ReadyProbes™ Reagent (Thermo Fisher, Waltham, MA) was applied to stain the nuclei of whole cells and the dead cells were stained by diluting PI (Life Technologies) to 1 g/mL in PBS. The cells were incubated at 37 °C in an incubator for 20 min, was gently pressed with a coverslip to convert the 3D spheroid into a 2D cell layer for visualization, and further incubated for 20 min. The viability of cells was confirmed via fluorescence microscope imaging. In the case of the conventional 2D culture dish, microfluidic channels and ECM-coated platforms, the culture medium was removed and the vessels were washed with phosphate buffered saline (PBS). The Calcein-AM (Life Technologies) and PI solutions, which were diluted to1 g/mL in PBS, were incubated at 37 °C for 20 min. Live and dead cells were confirmed by fluorescence microscopy.
Cancer cell culture in the microfluidic chip platform
To prepare a microchannel mold, SU-8 (MicroChem Corp., Westborough, MA) was patterned on a silicon wafer by photolithography and the prepared mold was replicated with the polydimethylsiloxane (PDMS) Sylgard 184 (Dow Corning, Midland, MI). In brief, the PDMS prepolymer and the crosslinker were mixed at a ratio of 10:1 and poured onto the microchannel mold. The bubbles were removed prior to curing in a vacuum desiccator and then the degassed PDMS was placed in an oven and allowed to cure at 80 °C for more than 2 h. The cured PDMS was peeled off from the mold and cut into pieces. For the complete microfluidic cell culture platform, the PDMS piece was treated with oxygen plasma and assembled with a glass slide. The microchannel was coated with poly-D-lysine hydrobromide (PDL; 0.1 mg/mL; Sigma-Aldrich, St. Louis, MO) solution for 3 h to allow cell adhesion. Later, the PDL solution was removed and the vessel was rinsed three times with PBS.
The brain tumor cell line T98G was removed from the culture dish and the cell density was adjusted to 105 cells/mL. The suspension was introduced into the microchannels. Immediately after dispensing, the flow was halted for 24 h so that the cells could adhere to the surface of the microchannel. To prevent the drying in the incubator, culture medium was added by inserting 1 mL tips containing medium at the inlet and outlet of the microchannels. After 24 h, a syringe pump was connected and shear stress of 0.1 dyn/cm2 was applied to the cells.
Cancer cell culture on the ECM-coated culture vessel
A fence made of PDMS was fabricated to contain the culture medium and it was bonded to the glass slide. To study the difference between the receptor-mediated binding and the surface charge-mediated binding, Matrigel (Dow Corning) and PDL solutions were applied to two glass slides at a concentration of 0.1 mg/mL and incubated for 3 h. After that, the glass slides were rinsed thoroughly as described above. T98G cells were dispensed (1 ml) into the two coated glass slides with the density adjusted to 105 cells/mL. The chips were kept in an incubator set to 5% CO2 and 37 °C and the culture medium was changed every three days.
DOX treatment of cancer cells cultured on the different platforms
To compare cellular sensitivity and resistance to drugs on different platforms, DOX was selected as a representative anticancer drug. DOX was first completely dissolved in dimethyl sulfoxide and diluted to a total concentration of 1 μg/mL in the culture medium or PBS. DOX-supplemented culture medium was introduced in each culture platform and incubated for three days and cell growth or changes induced by the drug were examined.
Cell growth rate and chemoresistance analysis
For the analysis of cell growth, the culture medium was removed from each platform and the vessels were washed with PBS and treated with 0.25% trypsin- Ethylenediaminetetraacetic acid (EDTA). The detached cells were recovered and counted. In the case of the 3D cancer spheroid, the total number of cells was directly counted and analyzed by Hoechst nuclear staining as described in the Section “Live-dead cell staining”. On the third day after DOX treatment, the effect of drug sensitivity was measured by collecting cells from each platform and determining the number of cells. In the case of the 3D spheroid cells, the effects of DOX were measured by two methods: (i) changes in the diameter of the spheroids and (ii) the ratio of PI-positive (dead) cells to the total cells (Hoechst fluorescence staining).
Results and discussion
To emulate the in vivo microenvironment of a native tumor with engineering techniques, three types of cell culture methods:—3D cancer spheroid culture, microfluidic chip culture for stimulation of shear stress by interstitial flow and ECM-coated surface culture for receptor-mediated adhesion—were utilized. The cellular behaviors, such as growth rate and chemoresistance, were compared with those of the conventional 2D monolayer cell culture models, and we characterized the changes of cells in each microenvironmental platform (Scheme 1). First, T98G cells were cultured on each platform to confirm the growth rate and viability. After the growth rates were compared, the changes in anticancer drug sensitivity in each environment were confirmed by treatment with DOX. This assay was designed to confirm the differences related to anticancer drug sensitivity between the culture models, thereby suggesting the requirements of an in vivo-like tumor model for the high-throughput screening of drug candidates.
Scheme 1. Culture of tumor cells on the tumor microenvironment-mimetic platforms. (A) The conventional 2 D monolayer cell culture method. (B) A 3 D spheroid cell culture method that can mimic the tight cell-cell interaction and high cell density of native tumors. (C) A method for culturing cells in the microfluidic channels capable of simulating a fluidic microenvironment in which an interstitial flow is present. (D) The culture of cancer cells in an ECM-coated substrate capable of reproducing the cell-ECM interaction between the receptors of cancer cells and the ligands of ECM matrices.
Comparative analysis of cancer cell growth on different platforms
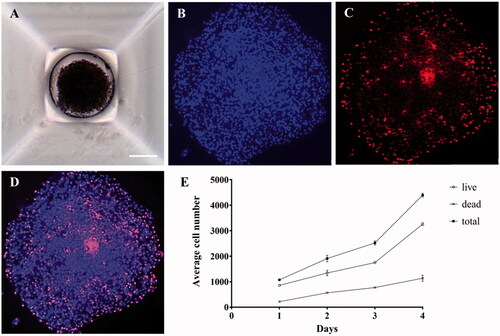
To confirm tumor cell growth in the 3D spheroid environment, T98G cells were cultured for four days in the microwells of SpheroFilmTM () as a spheroid form. The growth of cancer cells cultivated in the 3D environment can be confirmed by indirect methods such as an ATP assay or [3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay. However, in this study, embryo cell staining was performed for direct confirmation of 3D cell growth. After the immunohistochemical staining with Hoechst (), the nuclei of the grown cells were stained to count the whole population of cells, and the number of dead cells was determined by PI staining () to confirm the total cell growth rate and the percentage of dead cells (). Therefore, the viability can be determined by the ratio of the number of PI-positive cells (dead cells) to the number of Hoechst-positive nuclei (total cells). As a result, it was confirmed that after four days, the number of dead cells increased approximately fivefold, while the total number of cells increased approximately fourfold ().
Figure 1. Microscopic images of 3D cancer spheroids. (A) T98G cells were cultured with SpheroFilmtr™ to obtain a 3D spheroid form, and the appearance of spheroids was confirmed by optical microscopy (scale bar: 200 μm). (B–D) To count viable whole cells and dead cells in a 3D spheroid, the spheroid was transferred onto a glass slide, mounted with a coverslip, stained with Hoechst and PI, and examined under a fluorescence microscope. (B) Hoechst nuclear staining confirmed the total number of cells. (C) Dead cells were identified among whole cells by PI staining. (D) An overlay of Hoechst and PI staining images. (E) Analysis of viability of 3D spheroid cells by fluorescence staining as a function of culture time.
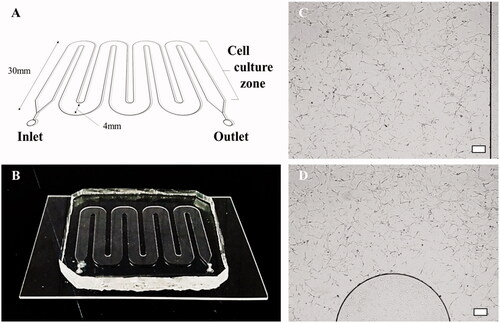
The cancer cells were cultured in a microfluidic chip () to identify the effect of fluidic shear stress on the cellular behaviors. To this end, the cells were initially introduced into the fluid channel and allowed to adhere to the chip for one day without applying culture media flow. From the second day, the cells were exposed to the fluidic condition at a constant flow rate using a syringe pump. The fluidic culture condition was adjusted to ensure shear stress of 0.1 dyn/cm2, which is comparable to the values observed in in vivo tumors (). After four days of cell culture, the cells were harvested by trypsin treatment and the number of cells was measured by a trypan blue dye exclusion assay. It was confirmed that the cell number increased by ∼15-fold as compared with the initial cell number.
Figure 2. Microfluidic chip and cell culture images in microchannels. (A, B) A schematic diagram of the microfluidic chip and an image of the chip fabricated with PDMS. (C, D) Micrographs showing that T98G cells grow well in the PDL-coated microfluidic chip (scale bar: 200 μm).
To confirm the growth of tumor cells in the presence of ECM, 105 cells were cultured in pretreated PDL-coated chips and Matrigel-coated chips (). In both PDL- and Matrigel-coated chips, cells showed an approximately 15-fold increase in cell number as compared with the initial number of seeded cells ().
Figure 3. The chip for comparison of receptor- and charge-mediated adhesion of cancer cells. (A) Photograph of the wells made of PDMS to contain PDL, Matrigel solutionand culture media. The well was bonded with a glass slide to prevent potential leakage of solutions. (B) Micrographs of T98G cells cultured on a PDL-coated chip and (C) T98G cells cultured on a Matrigel-coated chip (scale bar: 200 μm).

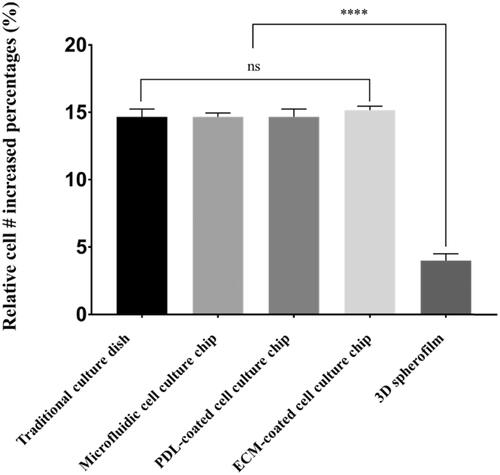
Overall, when tumor cells were cultured in the platforms coated with PDL and Matrigel, they increased in number by approximately 15-fold over the same incubation period, which was similar to the results obtained from the microfluidic culture platform. This finding was also similar to the results of cells cultured on a traditional 2D platform. Nevertheless, when the cells were cultured in a 3D spheroid form, the number of cells increased approximately fourfold during the same incubation period and the growth rate was remarkably slow as compared with other culture platforms ().
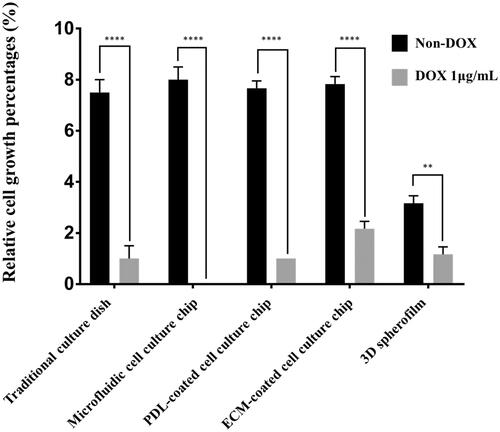
Figure 4. A comparison of cell growth rates depending on the culture platform. In the traditional 2D culture dish, microfluidic culture chip and ECM-coated platform, cells expanded by ∼15-fold in four days, while the 3D spheroids showed only a fourfold increase in cell number. These results demonstrate that the culture dimension (2D or 3D) can significantly affect the expansion rate of cancer cells. (****: p ≤ .0001)
The difference in growth rate seems to originate from the culture dimension. In the case of 2D-based culture models in which cells are cultured in a monolayer form, cells are consistently exposed to the culture media and thereby have unlimited access to oxygen and nutrients [Citation27]. These monolayer-based culture platforms include plastic culture dishes, microfluidic chips and ECM-coated culture vessels. On the other hand, the 3D cancer spheroids do not have sufficient access to nutrients and oxygen due to the high cell density. Indeed, the native tumor mass and the tumor spheroid have multiple distinct layers depending on the distance from the outer layer: a proliferating outer layer in which zone cells proliferate aggressively, a quiescent zone in which zone cells neither proliferate nor die and a necrotic core where cells are dying because of the limited oxygen and nutrient transport [Citation28]. In the case of the tumor spheroid, proliferation can occur only in the thin outer cell layer and thus the growth rate of the 3D spheroid is slower than that of other cell monolayer-based culture models.
Chemoresistance against the antitumor drug
After comparing the cell growth rates, we conducted experiments to compare the sensitivity and resistance of the cancer cells to DOX. When cells were cultured for three days in the conventional 2D culture dish, PDL-coated and Matrigel-coated chips and the microfluidic chip without DOX treatment, the cells expanded approximately sevenfold in number, while the 3D spheroid-cultured cells showed only a fourfold increase in cell number. However, when cultured with DOX-incorporated media for three days, the cells cultured in the 2D culture dish, PDL-coated chip or 3D spheroid film showed no changes in cell number, indicating an efficacy in the suppression of cancer growth (). By contrast, in the case of the Matrigel-coated chip, the number of cells increased by 2.5-fold over three days, even in the presence of DOX, although the growth rate was slower than in the non-treated case (sevenfold increase in the non-treated case), demonstrating a chemoresistance against DOX ().
Figure 5. Changes in cell growth on each platform after DOX treatment. The growth of T98G cancer cells with and without DOX treatment. When treated with 1 μg/mL DOX, the cells cultured on the 2 D culture dish, PDL-coated microfluidic chip, and 3D spheroid film did not show growth, whereas T98G cells cultured on the Matrigel-coated chips showed marginal growth of 2.5-fold. In addition, when shear stress was administered in the presence of DOX, the cells lost their adhesion and were completely washed out. (**: p ≤ .01, ****: p ≤ .0001)
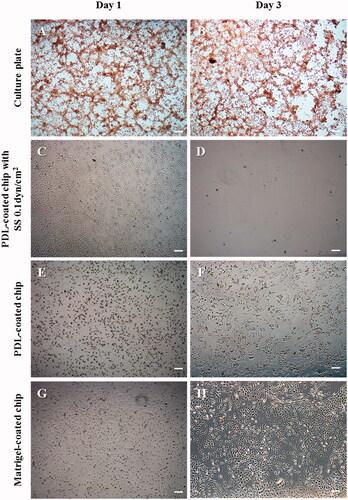
Furthermore, when the cancer cells were cultured with DOX in a 2D culture dish or PDL-coated chip, which are static culture conditions, the cells showed aberrant morphology, such as limited cell spreading area and loss of lamellipodia and filopodia; however, the cell number did not change at the early stage. (). In addition, after three days of incubation, the cells were washed out during exchange of the culture medium because of their weakened binding ability.
Figure 6. The morphological changes of T98G cells treated with 1 μg/mL DOX on the different cell culture platforms: a 2D cell culture plate, shear stress (SS) chip, PDL-coated chip or Matrigel-coated chip. When DOX treatment was administered, the cells did not proliferate in the culture dish; the lamellipodia and filopodia were not protruding from the cell body and the cell morphology changed to an aberrant form. In the microfluidic chip coated with PDL, T98G cells were almost completely washed out. This result suggests that the cells that lost their binding ability were separated by the flow. On the other hand, despite the DOX treatment in the Matrigel-coated chip, the morphology was unchanged and cell proliferation was observed (scale bar: 200 μm).
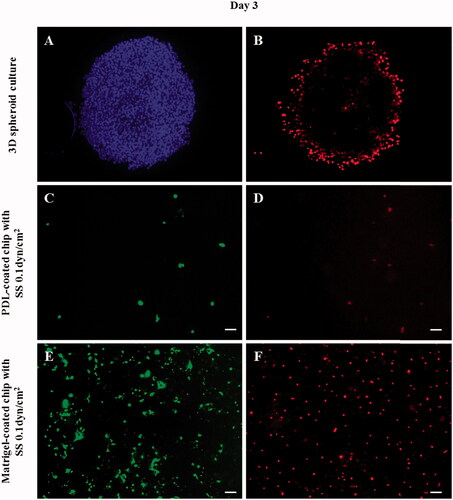
In the case of the PDL-coated microfluidic chip, the cells disappeared when they were incubated for three days under the fluidic shear stress condition (). This finding indicates that cells lost the binding ability under the influence of DOX and were washed out by the flow of the culture media (). On the other hand, despite the DOX treatment in the Matrigel-coated chip, the morphology was unchanged and cell proliferation was observed. The fluorescent staining confirmed that the cells were well bound to the surface of the chip and proliferating even during the continuous flow of culture media (). In the case of 3D culture, the appearance of viable tumor cells in the submarginal region of the spheroids was observed (no effect of DOX; ). Although it was not clear whether the underlying mechanism was due to cell–cell interactions or tight junction, the effect of DOX was only limited to the outer layer of the tumor cell spheroid (high concentration of PI-positive cells at the boundary).
Figure 7. Fluorescent images of cancer cells in the 3D spheroid platform and in a microfluidic chip after DOX treatment. According to the fluorescent images, cancer cells in the 3D spheroid form showed a limited effect of DOX as confirmed by the increased cell number and PI-positive cell number. PI staining (B,D,F: dead cells) confirmed that only the cells of the outer layer of the 3D spheroid were dying, indicating that only the surface cells exposed to DOX were affected, whereas the interior cells were not affected. Additionally, in the presence of flow in the PDL-coated microfluidic channel, the cells were washed out. On the other hand, in the Matrigel-coated channel, cells demonstrated a relatively strong resistance to DOX even in the presence of flow. (A: nuclei/Hoechst, B,D,F: dead cells/PI, and C,E: live cells/Calcein-AM). (scale bar: 200 μm).
The PDL coating modifies the surface charge of the culture platform into a positive form and promotes the electrostatic interaction between the positively-charged surfaces and the negatively-charged cell membrane. By doing so, the cells can attach to the culture vessel [Citation29]. On the other hand, the ECM-coated surface can present surface ligands to which the receptors in the cell membrane, such as integrin, can bind and thereby mediate the cell-ECM interaction. Such– ligand-receptor binding via cell-ECM interaction can “turn on” the intracellular signaling and modulate various cellular behaviors [Citation29,Citation30] that are not triggered by electrostatic interaction. Especially in the case of cancer, it is known that the cell-ECM interaction can induce cell adhesion-mediated drug resistance (CAM-DR). For example, Matsunaga et al. [Citation31] have demonstrated that the interaction of β1 integrin on leukemia cells with fibronectin can promote the acquisition of chemoresistance. Matrigel is a mouse sarcoma-derived ECM matrix and is known to contain various types of ECM components including laminin, collagen IV, heparin sulfate proteoglycans and entactin/nidogen, as well as a number of growth factors [Citation32]. Therefore, the cell-ECM interaction could induce chemoresistance against DOX when T98G cells were cultured in Matrigel-coated substrates ().
In the case of the 3D cancer spheroids, the dense packing of the cells can induce the following effects. First, the tight cell-cell interaction and high cell density can hinder the molecular transport of drugs into the deep core. Previous studies indicated that spheroid cultures are characterized by their limited drug and nanoparticle penetration compared with monolayer cultures [Citation33–38]. Representatively, a study using fluorescent nanoparticles demonstrated the accumulation of nanoparticles only in the outer cell layer [Citation39,Citation40]. Second, the hypoxic environment in the quiescent zone (submarginal zone between the outer cell layer and the necrotic core) can induce chemoresistance. For example, the expression of hypoxia-inducible factor-1 (HIF-1) caused by limited oxygen supply is related to the chemoresistance in multiple types of cancer cells [Citation41]. Third, the tight junction expression between the cells in the tumor spheroids may be involved in the different drug responses. Some researchers proposed that the tight junction mRNA may be involved in the acquisition of multi-drug resistance in gastric cancer [Citation42]. Although further studies are needed to identify the factors that mainly regulate drug responses, it is evident that the preparation of test models can significantly influence the drug screening outcomes.
Conclusions
Although numerous laboratory animals have been sacrificed for the fundamental study of human diseases or preclinical testing of new drugs, it has been known that animal experiments cannot predict outcomes in human clinical trials [Citation43,Citation44]. In addition, in vitro cell culture-based experiments involving conventional plastic culture dishes do not yield results that are similar to those in vivo [Citation45–47]. For these reasons, the drug discovery pipeline requires excessive time and resources to develop new drugs [Citation48] and experimental platforms that can predict efficacy and cytotoxicity similar to those in clinical trials are urgently needed. In order to overcome the limitation and problem of conventional 2D monolayer cell culture platform, various cell culture platforms that address the physiology of the cancer microenvironment were prepared. We found tumor cell growth and chemoresistance were significantly different depending on the culture conditions such as the culture dimensions, fluidic shear stress and the presence of ECM. These results showed that the physiological characteristics of tumor microenvironment have to be considered to avoid potential misleading during anti-cancer drug tests or in vitro tumor experiments. It is believed that if multiple features of the tumor microenvironment are implemented on a single platform, the outcomes can indicate the efficacy of a drug in clinical trials. Further research and development would be needed to design more physiologically relevant cancer cell culture platform that is simple yet robust to use in the laboratory. Then, in the near future, in vitro cancer platform may replace animal experiments and we hope that they overcome the limitations of existing in vitro experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Rodriguez-Devora JI, Zhang BM, Reyna D, et al. High throughput miniature drug-screening platform using bioprinting technology. Biofabrication. 2012 Sep;4. DOI:https://doi.org/10.1088/1758-5082/4/3/035001
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22:151–185.
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33.
- Akhtar AZ, Pippin JJ, Sandusky CB. Animal studies in spinal cord injury: a systematic review of methylprednisolone. Altern Lab Anim. 2009;37:43–62.
- Burgon J, Hamerslag L. The relationship between scientific research, clinical trials and FDA drug approval. Value Health. 2013;16:A478–A47A.
- Taylor K, Gordon N, Langley G, et al. Estimates for worldwide laboratory animal use in 2005: authors' response. Atla-Altern Lab Anim. 2008;36:496–497.
- Hay M, Thomas DW, Craighead JL, et al. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51.
- DiMasi JA, Feldman L, Seckler A, et al. Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther. 2010;87:272–277.
- Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000-2009. Clin Pharmacol Ther. 2011;89:183–188.
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715.
- Akhtar A. The flaws and human harms of animal experimentation. Camb Q Healthc Ethics. 2015;24:407–419.
- Ledford H. Translational research: 4 ways to fix the clinical trial. Nature. 2011;477:526–528.
- Arrowsmith J. Trial watch: phase III and submission failures: 2007-2010. Nat Rev Drug Discov. 2011;10:87.
- Mak IWY, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014;6:114–118.
- Curt GA, Chabner BA. One in five cancer clinical trials is published: a terrible symptom-what's the diagnosis? Oncologist. 2008;13:923–924.
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose?. Nat Rev Drug Discov. 2014;13:497–512.
- Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–520.
- Stringfellow DA, Gray BW, Lauerman LH, et al. Monolayer culture of cells originating from a preimplantation bovine embryo. In Vitro Cell Dev Biol. 1987;23:750–754.
- Li Y, Killian KA. Bridging the Gap: from 2D cell culture to 3D microengineered extracellular matrices. Adv Healthc Mater. 2015;4:2780–2796.
- Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–373.
- Wu MH, Huang SB, Lee GB. Microfluidic cell culture systems for drug research. Lab Chip. 2010;10:939–956.
- Holle AW, Young JL, Spatz JP. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv Drug Deliv Rev. 2016;97:270–279.
- Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem. 2007;101:805–815.
- Landowski TH, Olashaw NE, Agrawal D, et al. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–2421.
- Mbeunkui F, Johann DJ. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol. 2009;63:571–582.
- Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912.
- Cox MC, Reese LM, Bickford LR, et al. Toward the broad adoption of 3D tumor models in the cancer drug pipeline. Acs Biomater Sci Eng. 2015;1:877–894.
- Mehta G, Hsiao AY, Ingram M, et al. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J Control Release. 2012;164:192–204.
- Chen QM, Kinch MS, Lin TH, et al. Integrin-mediated cell-adhesion activates mitogen-activated protein-kinases. J Biol Chem. 1994;269:26602–26605.
- Kim HN, Jang KJ, Shin JY, et al. Artificial slanted nanocilia array as a mechanotransducer for controlling cell polarity. ACS Nano. 2017;11:730–741.
- Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–1165.
- Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010;10:1886–1890.
- Perche F, Torchilin VP. Cancer cell spheroids as a model to evaluate chemotherapy protocols. Cancer. Biol Ther. 2012;13:1205–1213.
- Desoize B, Jardillier J. Multicellular resistance: a paradigm for clinical resistance? Crit Rev Oncol Hematol. 2000;36:193–207.
- El Bayoumil T, Torchilin VP. Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin Cancer Res. 2009;15:1973–1980.
- Le VM, Lang MD, Shi WB, et al. A collagen-based multicellular tumor spheroid model for evaluation of the efficiency of nanoparticle drug delivery. Artif Cells Nanomed Biotechnol. 2016;44:540–544.
- Safdar MH, Hussain Z, Abourehab MAS, et al. New developments and clinical transition of hyaluronic acid-based nanotherapeutics for treatment of cancer: reversing multidrug resistance, tumour-specific targetability and improved anticancer efficacy. Artif Cells Nanomed Biotechnol 2017 [Oct 30]:[14 p.]. DOI:https://doi.org/10.1080/21691401.2017.1397001
- Usmani A, Mishra A, Ahmad M. Nanomedicines: a theranostic approach for hepatocellular carcinoma. Artif Cells Nanomed Biotechnol. 2017 [Sep 8]:[11 p.]. DOI:https://doi.org/10.1080/21691401.2017.1374282
- Goodman TT, Olive PL, Pun SH. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int J Nanomedicine. 2007;2:265–274.
- Albanese A, Lam AK, Sykes EA, et al. Tumour-on-a-chip provides an optical window into nanoparticle tissue transport. Nat Comms. 2013;4:2718.
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410.
- Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788:872–891.
- Panksepp J. Affective preclinical modeling of psychiatric disorders: taking imbalanced primal emotional feelings of animals seriously in our search for novel antidepressants. Dialogues Clin Neuro. 2015;17:363–379.
- Akhtar A. Animals and public health: why treating animals better is critical to human welfare. Choice: Curr Rev Acad Libraries 2012;50:515.
- Greek R, Menache A. Systematic reviews of animal models: methodology versus epistemology. Int J Med Sci. 2013;10:206–221.
- Ehrmann RL, Gey GO. The growth of cells on a transparent gel of reconstituted rat-tail collagen. J Natl Cancer Inst. 1956;16:1375–1403.
- Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32:760–772.
- DiMasi JA, Hansen RW, Grabowski HG, et al. Cost of innovation in the pharmaceutical industry. J Health Econ. 1991;10:107–142.