
Abstract
Mesenchymal stem cells (MSCs) that display homing and infiltration properties towards tumor cells are a promising cellular targeting vector for brain tumor therapy but are limited to local-regional delivery in current preclinical models. Here, we investigated whether placenta-derived MSCs (P-MSCs) are a superior cellular vector for systemic targeting of glioblastoma stem-like cells (GSCs), with an imaging modality to real-time monitor the trafficking P-MSCs to glioblastoma sites. Results demonstrated that P-MSCs had greater migratory activity towards GSCs and across blood–brain barrier compared with bone marrow-derived MSCs, and this activity was enhanced by hypoxia precondition. Chemokine ligand 5 was identified as a chemoattractant responsible for the glioblastoma tropism of P-MSCs. Polyethylene glycol-coated superparamagnetic iron oxide (PEG–SPIO) was synthesized for cellular labelling and imaging P-MSCs, displaying high cellular uptake and no cytotoxic effect on P-MSCs cell proliferation or stemness property. The homing effects of intravenously administered PEG–SPIO-labelled P-MSCs towards intracerebral GSCs were able to be detected in mice models through T2-weighted magnetic resonance imaging (MRI). This study suggests the possibility of innovative systemic P-MSC-based cell therapy for aggressive GSCs, developing a state-of-the-art theranostic technique for real-time tracking of therapeutic P-MSCs tumor infiltration through cellular MRI.
Introduction
Glioblastoma is the most prevalent and lethal type of malignant glioma, but the outcomes of its standard treatments – including surgical resection, radiation and chemotherapy – remain poor with a median survival of only about 15 months following diagnosis [Citation1]. The fundamental problem of these malignancies is their highly infiltrative nature and extreme resistance to conventional treatments. Recently, the subpopulation of glioblastoma termed “glioblastoma stem-like cells” (GSCs) are found to retain many of the capabilities of normal stem cells—including self-renewal, high expression of drug-efflux pumps, high capacity for DNA repair and maintaining the neoplastic clone—and are thought to be responsible for glioblastoma tumor initiation, maintenance and recurrence [Citation2,Citation3]. The results of glioblastoma treatment are also limited by the blood–brain barrier (BBB), which regulates the delivery of molecules in the brain [Citation4], the high sensitivity of brain tissue that allows only limited doses of therapeutic agents to be employed and the absence of a sensitive imaging platform enabling early diagnosis [Citation5,Citation6]. Therefore, developing a theranostic agent with GSC targeting, BBB accessibility and monitoring abilities may help to manage or even to eradicate glioblastoma.
Developing an effective tool to specifically deliver GSC-targeted therapies is a formidable challenge. Recently, engineered neural stem cells (NSCs) employed as a tumor-targeting strategy have demonstrated an extensive homing effect that inhibits glioblastoma growth in experimental animal models [Citation7,Citation8]. Nevertheless, ethical and logistical problems are associated with engineering NSCs, which must be isolated from neonatal tissue and require immunologic compatibility for allogenic transplantation; thus, identifying replacement stem cells for clinical applications is warranted [Citation9]. When participating in wound repair, mesenchymal stem cells (MSCs) exhibit high tropism towards sites of injury, inflammation and neoplasms [Citation10–12]. Researchers conducting clinical trials that have used systemic infusion of MSCs for treating neurological diseases caused by injury or stroke have speculated that exogenous MSCs can migrate across the BBB [Citation13,Citation14]. Shi et al. [Citation15] suggested that tumors can be considered as wounds that never repair, and continuous recruitment of MSCs may become vital for the delivery of tumoricidal agents. Remarkably, bone marrow-derived MSCs (BM-MSCs) have been shown to possess extensive tropism and homing towards brain tumors when administered intracranially [Citation16]. Although bone marrow is the traditional source of human MSCs, the placenta has recently emerged as an alternate and attractive source for MSCs because it provides a large volume of cells that can be widely acquired with minimal ethical controversy [Citation17]. Placenta-derived MSCs (P-MSCs) have exhibited superior biological cell properties compared to BM-MSCs, including higher expansion and engraftment capacities [Citation18,Citation19]. As such, P-MSCs are used in a broad range of clinical applications covering regenerative medicine and the treatment of inflammatory diseases and immune disorders [Citation20–22]. Moreover, emerging studies validate that P-MSCs either unmodified [Citation23] or carrying therapeutic genes [Citation24,Citation25] have antitumor activity, further implying that P-MSCs may be a superior stem cell type over NSCs and BM-MSCs for clinical applications targeting glioblastoma.
Development of stem cell-based therapy requires monitoring the fate and distribution of transplanted stem cells to maximize the therapeutic benefit. Hence, their accurate detection and localization in real time using clinically relevant imaging techniques is essential. Because of its non-invasiveness, high spatial resolution, three-dimentional acquisition and serial monitoring, magnetic resonance imaging (MRI) has become the preferred imaging technique for precise tracking of cell migration and the homing effect in living organisms [Citation26–28]. Superparamagnetic iron oxide (SPIO) nanoparticles are recently employed as cellular MRI probes for clinical application [Citation29,Citation30], which allows labelled cells to be tracked by T2 MRI under micromolar concentrations of iron [Citation31]. Study results have suggested that the uptake of SPIO nanoparticles spontaneously phagocytosed in different types of MSCs provides an exceptional in vivo tool by which MRI can monitor MSCs dynamically [Citation32,Citation33]. To prevent destabilization of the colloidal suspension and to make SPIO nanoparticles soluble in biological media, hydrophilic coating of SPIO nanoparticles has been recommended. Polyethylene glycol (PEG), a glycol non-ionic surface-active agent, is the most widely used biocompatible polymer for this purpose. PEGylation is shown to enhance the hydrophilicity, dissolubility and circulation time and reduce the toxicity and immunogenicity of the conjugated nanoparticles, and the technique has been FDA-approved for use in various drug formulations [Citation34,Citation35].
To translate the benefits of MSCs to clinical use, this study aimed to (1) compare the glioblastoma tumor-homing activity of MSCs derived from bone marrow and placenta, and augment their motility using an adaptive strategy; (2) employ PEG-SPIO nanoparticles for effective labelling and tracking of P-MSC and (3) establish a non-invasive MRI-based platform for real-time monitoring of the systemically transplanted P-MSCs tumor infiltration and distribution patterns in clinically relevant glioblastoma mouse models. This study provides essential information for GSC target-site delivery of MSC-based therapeutics with useful clinical applications.
Materials and methods
Cell lines and cell culture
Human BM-MSCs were purchased from Lonza (Rockland, ME) and maintained in MSCGM™ according to the manufacturer’s instructions (Lonza). Human P-MSCs stably transfected with green fluorescent protein and firefly luciferase genes (GFP-Fluc P-MSC) and corresponding growth media were provided by Meridigen Biotech Co., Ltd (Taipei, Taiwan). All MSCs used in this study were between the fourth and sixth passage. Human glioblastoma U87 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in high-glucose Dulbecco's modified essential medium (DMEM-HG; Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Genedirex, Las Vegas, NV). U87 GSCs were generated under the tumor sphere culture condition by plating cells in 24-well ultra-low-attachment plates (Corning Incorporated, Corning, NY) with DMEM/F12 serum-free medium supplemented with 20 ng/mL human recombinant epidermal growth factor (Sigma-Aldrich, St. Louis, MO), 20 ng/mL basic fibroblast growth factor (Sigma-Aldrich) and 1× B27 (Invitrogen). For the hypoxia preconditioning, MSCs were incubated in a hypoxic chamber (1% O2, 4% CO2 and 95% N2) for 2 or 6 h.
Osteogenic and adipogenic differentiation
For osteogenic differentiation, P-MSCs were seeded and incubated in a 6-well plate at a density of 104 cells/cm2 with DMEM-HG with 10% FBS, 10 mM β-glycerol-phosphate, 50 mM ascorbate-2-phosphate and 100 nM dexamethasone. For adipogenic differentiation, P-MSCs were incubated with DMEM-HG containing 10% FBS, 1 μM dexamethasone, 0.5 mM methylisobutylxanthine and 0.1 mM ascorbic acid. The medium was changed twice a week for 2 weeks, and cells were subjected to Alizarin Red S staining for osteogenic differentiation or Oil Red O staining for adipogenic differentiation using commercial staining solutions. To quantify staining, dyes were extracted from cells with isopropanol containing 4% Nonidet P-40 (Sigma-Aldrich) and measured as absorbance at wavelengths of 550 and 500 nm. All reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Transwell migration and in vitro BBB assay
A total of 105 U87 or U87 GSCs were grown in the lower chamber of Transwells (EMD Millipore) containing RPMI 1640 medium (Invitrogen) under the serum-free condition. After 3 h of incubation, 105 P-MSCs or BM-MSCs in 200 μL of serum-free RPMI medium were added to the upper chamber containing 8-μm-pore polycarbonate. MSCs that migrated through the membrane were stained with 0.5% crystal violet. Random fields (five per membrane) were photographed at 10× magnification and then quantified by measuring the absorbance of dye extracts at 570 nm. For in vitro BBB assay, a model consisting of rat brain capillary endothelial cells, brain pericytes and astrocytes was established using a ready-to-use BBB kit following the manufacturer’s instructions (PharmaCo-Cell, Nagasaki, Japan). The BBB kit was incubated at 37 °C in 5% CO2 for 4 days allowing for functional activation. P-MSCs and BM-MSCs with and without the hypoxia precondition were labelled with Vybrant carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) cell tracer (Invitrogen) prior to the migration assay according to the manufacturer's protocol. The 105 CFDA-labelled MSCs in 200 μL of serum-free RPMI medium were added to the top of the BBB insert, and 1 ml of 5% FBS in RPMI medium was added to the bottom chamber of the transwells. After 9 h of incubation, the entire transmigrating cell population present in the bottom chamber was counted under a Zeiss Axiovert 200 fluorescent microscope (Zeiss, Salt Lake City, UT) with a 10× objective. Each experiment was performed three times.
Chemokine membrane arrays and ELISA
The experimental procedure followed the manufacturer’s instructions for the human chemokine antibody array C1 (AAH-CHE-1; RayBiotech, Inc., Norcross, GA). Conditioned media acquired from U87 cells and U87 GSCs were applied to the membranes for 2 h. After washing, array antibodies and horseradish peroxidase–conjugated streptavidin were incubated with the membranes for 2 h. The membranes were then added to 1× detection buffers, and photographs were obtained using a charge-coupled device camera. The reagent of the human CCL5 ELISA kit was used (R&D Systems, Minneapolis, MN), and the expression test was performed strictly according to the instructions.
Synthesis of PEG–SPIO and cell labelling
SPIO nanoparticles enclosed by oleylamine were synthesized through thermal decomposition [Citation36]. Initially, oleylamine (10 ml, >50% purity, TCI), tris (acetylacetonato) iron(III) (1 g, abb. Fe(acac)3, Acros Organics) and diphenyl ether (10 ml, 99%, Acros Organics) were degassed at 110 °C under nitrogen for 1 h. The mixture was then heated to 300 °C, and SPIO nanoparticles with oleylamine surface modification were formed through thermal decomposition under a nitrogen atmosphere for 1 h. The oleylamine-coated SPIO were separated through magnetic separation. Subsequently, PEG silane (2-[methoxy(polyethylenoxy)propyl] trimethoxysilane, 0.6 ml, 1% v/v, Sigma-Aldrich) was added to the oleylamine-coated SPIO (with a density of 0.6 g/mL in 10 ml of hexane) for ligand exchange [Citation37] with the aid of acetic acid (2.5 ml, 0.05% v/v, Sigma-Aldrich). Finally, water-soluble PEG–SPIO (with a density of 1.08 g/mL in 2 ml of water) was extracted from the mixture through magnetic separation that was incubated in a shaker at room temperature for 24 h. For the cell labelling, P-MSCs grown on culture plates were replaced with fresh medium containing various concentrations of PEG-SPIO (0–120 μg/mL) at 37 °C for 30 min, and then placed on the magnetic plate (OZ biosciences, Marseille, France) for 20 min. Cells were washed three times with phosphate buffered saline (PBS) to eliminate extracellular PEG-SPIO, and then harvested for the additional assays.
Prussian blue staining and transmission electron microscopy (TEM)
Cellular uptake and localization of the SPIO particles in P-MSCs was observed through Prussian blue staining and TEM. A total of 5 × 104 PEG–SPIO-labelled P-MSCs were seeded and incubated in a 24-well plate for 24 h, and then subjected to ion staining using a Prussian Blue staining kit according to the manufacture’s protocol (Ocean NanoTech, Springdale, AR). After counterstaining with nuclear fast red, cell images were captured under microscope with a 10× objective. For the TEM, PEG–SPIO-labelled P-MSCs grown on chamber slides were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 30 min at room temperature, and then subjected to post-fixation with 2% osmium tetroxide. After dehydration in ascending grades of ethanol and propylene oxide, samples were embedded with Epon and polymerization at 62 °C for 48 h. Ultrathin sections (∼70 nm) were obtained using an ultramicrotome (Leica Ultracut UCT, Leica Microsystems GmbH, Wetzlar, Germany) and collected on 100 mesh copper grids. TEM images were captured using a Hitachi HT-7700 electron microscope (Hitachi High-Tech, Tokyo, Japan) operating at an 80–200 kV accelerating voltage.
In vitro cellular MRI
Different concentrations of PEG–SPIO (0–75 μg/mL) were prepared as standard phantoms to validate the maximal visible concentration of iron. PEG–SPIO-labelled (0–45 μg/mL) and unlabelled P-MSCs were trypsinized, centrifuged and resuspended in 0.5 ml of 1% agarose in Eppendorf tubes. Contrast-enhanced MRI of cells was performed using a 7 T Small-Animal MRI System (Bruker Biospec, Ettlingen, Germany). The operational software of the scanner was Paravision PV6.0.1 (Bruker). For T2 curve fit evaluation, a multislice multiecho sequence was acquired using the following parameters: repetition time (TR), 3000 ms; echo time (TE), 8.5–204 ms; echo spacing, 8.5 ms; flip angle, 90°; matrix, 256 × 256 interpolated; field of view, 42 mm; echo train length, 8 and slice thickness, 0.5 mm, with 16 slices taken. The signal decay rates for all echo times were assessed [Citation38].
Animal study
All animal experiments were approved by the Institutional Animal Care and Use Committee of Taipei Medical University (LAC-2014–0156) and China Medical University (104–25-N) and complied with their regulations. For the establishment of brain tumors in orthotopic models, animals (BALB/c nude) were anesthetized by intraperitoneal (IP) administration of tiletamine (Zoletil; 25 mg/kg) and xylazine (Rompun; 12.5 mg/kg). The heads of the recipient mice were secured in a stereotactic frame (Stoelting Co., Wood Dale, IL). A burr hole was prepared 0.5 mm posterior to the bregma and 2.5 mm to the right of the sagittal suture using a microdrill. U87 GSCs in 3 μL were slowly injected using a syringe (Hamilton, Bonaduz, Switzerland) at 2 mm below the brain surface. The syringe remained in place for 5 min prior to withdrawal. The skin was closed with an Ethilon 3–0 suture (Ethicon, Somerville, NJ). The sham control group consisted of two animals that received PBS without cancer cells. A third mouse with no treatment was used as a blank control. For MSC tracking, PEG-SPIO-labelled GFP-Fluc P-MSCs were hypoxia-preconditioned for 6 h and then administered into normal mice (n = 3 via each injection route) through intravenous (IV) or IP injection or into tumor-bearing mice (n > 5 each tumor type) through IV injection 2 weeks after cancer cell implantation. The mice were then imaged as described in the following sections.
Bioluminescence imaging (BLI)
In vivo BLI was conducted on a cryogenically cooled IVIS 200 system (Caliper Lifesciences, Hopkinton, MA) using Living Imaging acquisition and analysis software (Caliper Lifesciences). Mice were anesthetized with 5% isoflurane (Abbott, North Chicago, IL) and subsequently received an IP injection of 25 mg/mL of luciferin potassium salt (Gold Biotechnology, St. Louis, MO) in PBS at a dose of 125 mg/kg. The animals were then placed in a light-tight chamber; anesthesia was continued with 2% isoflurane introduced via a nose cone. Images were acquired 15 min after luciferin administration. An integration time of 1 min with a binning of 100 pixels was used for luminescence image acquisition in both the dorsal and ventral positions. Signal intensity was quantified as the sum of all detected photon counts within the region of interest after subtraction of the background luminescence.
In vivo MRI
MRI was performed using a Biospec 70/16 spectrometer equipped with a 7-T 40-cm magnet, a Biospec Bruker console and a surface coil (2.3-cm ID) for brain imaging. T2 rapid acquisition with a relaxation enhancement (RARE) sequence was used to examine the distribution of transplanted MSCs at 0, 4 and 7 days after MSC transplantation. The RARE parameters were as follows: TR, 2500 ms; TE, 33 ms; flip angle, 90°; matrix, 256 × 256 interpolated; field of view, 42 mm; echo train length, 8 and slice thickness, 0.5 mm, with 16 slices. Prior to MRI, mice were anaesthetized with isoflurane administered via a face mask and placed in a cradle made of Plexiglas. The head was fixed with a tooth holder. Body temperature was maintained at 37 °C ± 1 °C using warming blankets or integrated water hoses in the animal beds. Respiration and body temperature were monitored throughout the imaging.
Polymerase chain reaction (PCR)
The genomic DNA of frozen brain tissues was isolated using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The DNA was quantified with the use of a NanoDrop2000 spectrophotometer (Thermo Scientific). PCR amplification was performed with 10 ng and 100 ng of genomic DNA derived from cell lines and mouse brains, respectively. The primers used for PCR were as follows: Luc (luciferase-specific) forward-as: 5′-GCCAAAAACATTAAGAAGGGC-3′ and reverse-as: 5'- ATGTCCACCTCGATATGTGC-3′; CISD2 (mouse-specific) forward-as: 5′-CTCGGAGGTGAGTAGTCTAGAG-3′ and reverse-as: 5′-CGTCAGCTACAGTGCTTTCTAATCTC-3′. The cycling parameters were initial denaturation at 95 °C for 5 min followed by 35 cycles, with each cycle consisting of denaturation at 94 °C for 30 s, 55 °C for 15 s and 72 °C for 30 s; a final extension was then performed at 72 °C for 7 min. PCR products were separated using 1.5% agarose gel electrophoresis.
Statistical analysis
Statistical analysis was performed using Excel 2017 (Microsoft, Redmond, WA). Differences between the two groups were measured using the Student’s t test. Data are represented as mean ± standard error, and differences were deemed to be statistically significant at P < .05.
Results and discussion
Hypoxic preconditioning enhances P-MSC migration towards glioblastoma cells and capacity for transmigration across the BBB
We examined and compared the glioma-homing properties of P-MSCs and BM-MSCs in an in vitro co-culture cell model in which human glioblastoma U87 cells, U87 stem cell-like cells (U87 GSCs) or plain culture medium were placed on the bottom chamber of transwells and P-MSCs or BM-MSCs were seeded on the top chamber. After 3 h of incubation, no significant differences were found in the basal level of migration between the two MSC lines; however, the motility of the P-MSCs towards U87 and U87 GSCs was significantly higher (∼40% higher) than that of the BM-MSCs (). Recently, various in vitro preconditioning strategies have been suggested to improve MSC function. Hypoxic preconditioning was demonstrated to enhance migration/homing activities of BM-MSCs [Citation39] but has not yet been investigated in P-MSCs. To determine whether there was a significant difference between normoxic and hypoxic preconditions for GSC-induced MSC migration, a hypoxia chamber with 1% oxygen was used for cell culture. A time-dependent significant increase in migration was observed in the P-MSCs that were precultured under hypoxic conditions, but post-hypoxia did not affect cell migration (). Subsequently, to further identify the chemoattractive factors that promote the migration of P-MSCs to glioblastoma cells, a human chemokine antibody array was used to compare the chemokine expression profiles of the conditioned media collected from U87 cells and U87 GSCs. A total of 37 cytokines were tested, and expression of CCL27, CCL3, CCL4 and CCL5 was higher in U87 cells than in U87 GSCs (). Among these differentially expressed chemokines, CCL5 (alternatively named RANTES) has been reported to correlate with tumor progression and the promotion of immune cell infiltration in several cancers, including glioblastoma [Citation40,Citation41]. Interestingly, chemokine receptor profiling shows that BM-MSCs express the CCL5 receptors CCR1, CCR3 and CCR5 [Citation42,Citation43], and the chemokine signaling via CCL5 and its receptors is pivotal for the recruitment of BM-MSCs towards brain lesions [Citation44]. In addition to antibody array, we validated that U87 cells secreted an approximately ninefold higher amount of CCL5 than did U87 GSCs through enzyme-linked immunosorbent assay (); this result correlated with the higher ability of U87 than that of U87 GSCs to attract MSC migration seen in co-culture. Moreover, exogenous human CCL5 induced the transwell migration of P-MSCs (). Collectively, these results imply that CCL5 is, at least in part, a glioma-derived chemotactic factor responsible for MSC tumor tropism.
Figure 1. Migration ability of MSCs towards glioblastoma cells and across the BBB in vitro. BM-MSCs and P-MSCs that were preincubated under (A) normoxia or (B) hypoxia (1% O2) condition for 2 and 6 h were seeded on the top of transwells to allow migration towards the bottom wells containing U87, U87 GSC, or plain medium for 3 h. Migrated MSCs were stained with 0.5% crystal violet and photographed at 100× magnification. Numbers of migrated cells were quantified by measuring the absorbance of a dye extract at 570 nm. Assays were performed in triplicate in two independent experiments. Data are presented as mean ± SD of one representative experiment. a1 represents P ≦ .05 compared with BM-MSCs. b1 represents P ≦ .05 compared with RPMI medium. (C) The conditioned media from U87 and U87 GSCs were extracted, and human chemokine antibody array was performed. The differential expressed chemokines are indicated with rectangular boxes. (D) The expression level of CCL5 was quantified by ELISA; **P ≦ .005 compared with U87 GSC conditioned medium. (E) Chemotactic response of P-MSCs induced by human recombinant CCL5; *P ≦ .05 compared with serum-free medium alone. (F) The entire transmigrating cell populations were assayed using a BBB kit, photographed at 100× magnification, and counted using Image J software. a2 and c2 represent P ≦ .01 compared with BM-MSCs and RPMI, respectively. All assays were performed in two independent experiments in triplicates. Data are presented as the mean ± SD of one representative experiment.
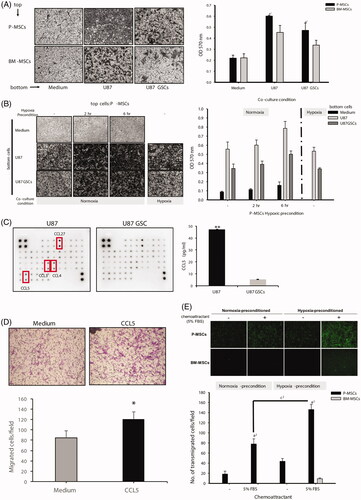
The BBB is a major hurdle in delivering drugs into brain tumors successfully, thus we assessed the effects of hypoxic preconditioning on MSC transmigration across the BBB using a ready-to-use in vitro BBB kit. We discovered that the BM-MSCs had almost no effect on passive diffusion across the BBB and that this activity was induced in the presence of chemoattractant (5% FBS) only when the cells were incubated in hypoxia before being placed in the testing system. In contrast, the P-MSCs were capable of migration across the BBB regardless of the external chemoattractant, and hypoxic preconditioning of the P-MSCs led to a twofold increase in both the basal level and chemoattractant-induced BBB transmigration (). The intrinsic mechanisms governing the differences in the migration activity of MSCs derived from different sources or with/without hypoxic stimulation remain to be determined. Previous studies have detected no significant difference in the expression levels of cell-surface antigens and chemokine receptors between P-MSCs and BM-MSCs [Citation45,Citation46]. However, some proteolytic enzymes – such as lysosomal proteases cathepsin B and cathepsin D [Citation47] and matrix metalloproteinases (MMPs) MMP-1 [Citation48], MMP-2, MT1-MMP and TIMP-2 [Citation49] – have been identified as being involved in BM-MSC migration, and most of these proteases are also hypoxia-induced genes. Comparing the expression level of these proteolytic enzymes for further mechanistic validation is warranted in the near future.
The MSCs that displayed the highest transmigration activity in both the tumor-homing and BBB models were found to be the hypoxia-preconditioned P-MSCs. Therefore, we used the P-MSCs that were pretreated with 6 h of hypoxia for the remainder of the experiments.
Intravenously transplanted P-MSCs are rapidly cleared in healthy non-tumor-bearing mice
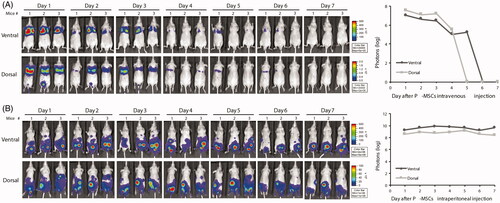
Regional MSC delivery by either direct injection into the cerebral hemisphere or intra-arterial injection via the internal carotid artery, as previously described [Citation50–52], achieves a relatively high cerebral engraftment rate in most experimental glioma therapies but requires invasive surgery or causes serious cerebrovascular complications such as microembolic strokes [Citation53,Citation54]. To determine the fate of systemically administered P-MSCs in homeostatic mouse recipients, two frequently used medication delivery routes, intravenous (IV) and intraperitoneal (IP) injection, were chosen for systemic delivery of P-MSCs that express green fluorescent protein and firefly luciferase into normal nude mice. This procedure was followed by BLI to track the location of the P-MSCs over time. Two distinct hotspots of activity were observed for mice receiving IV P-MSCs (); these hotspots corresponded to the location of the two lung lobes at 1 day after injection. The bioluminescent signal was retained, localized to the lungs, and gradually decreased with time (, imaging). At 5 days post-injection, the photon count over the whole body had lowered to a level undetectable using the default software (, plot). P-MSC activity in the lung also disappeared within 7 days. Imaging of mice receiving IP GFP-Fluc P-MSCs showed bioluminescence signals in the lower abdomen (, imaging). Light emission was sustained with no significant decline in photon count for at least 7 days (, plot). This result demonstrated a rapid clearance of IV P-MSCs in healthy, non-tumor-bearing mice, suggesting a safe dosing route for systemic P-MSC-based cell therapy.
Figure 2. Biodistribution of transplanted P-MSCs in homeostatic animals through different administration methods. Ventral and dorsal views of BLI of healthy, non-tumor-bearing mice receiving luciferase-expressing P-MSCs (GFP-Fluc P-MSC) through (A) IV or (B) IP injection (n = 3). Images were taken daily after P-MSC administration. Signals were adjusted to the same color scale for each image. Quantitative data are presented as means of logarithmic values of the whole-body total photon counts.
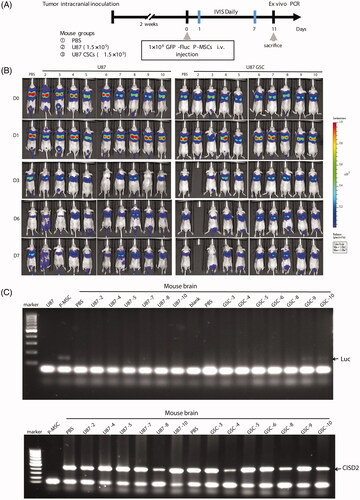
In vivo glioblastoma stem-like environment effectively triggers P-MSC infiltration
To validate whether the stemness condition of glioblastoma may alter the infiltration ability of P-MSCs, U87 and U87 GSCs were used in the animal model to investigate the recruitment of hypoxia-preconditioned P-MSCs after IV injection; the animal experimental procedure is shown in . The bioluminescent signal was retained, localized to the lungs, and gradually decreased with time in both the glioma-bearing mice (U87 and U87 GSCs) and sham control mice (phosphate-buffered saline) receiving P-MSCs. No visible light emission was observed at the site of brain tumor formation over 7 days of monitoring (). Considering the sensitivity of whole-body imaging, we collected brain tissues and performed polymerase chain reaction (PCR) analysis for luciferase gene expression. Among the brains extracted from the mice administered with P-MSCs, 100% of the sham control (2/2) and U87 (6/6) groups showed no detectable luciferase gene in their entire DNA. In contrast, the PCR product of the luciferase gene was seen in 2/7 (28.6%) brain tissues of the U87 GSC tumor-bearing mice after 35 cycles of amplification (). Although the positive rate was not high using the present detection modality, this result demonstrated the potential of P-MSCs to migrate to intracranial glioblastoma containing stem-cell-like malignant cells through IV administration. U87 GSCs were found to express a lower amount of CCL5 and were less efficient at triggering MSC migration in cell culture (), but they exhibited higher potential for recruiting systemically administered MSCs to the tumor sites than their monolayer parental cells when the cells were implanted into mouse brains. An explanation for this could be the tumor-initiating property of GSCs, which slows the cell proliferation in vitro but increases the number of U87 GSCs after transplantation by accelerating both tumor engraftment and the tumor growth rate. Additionally, microenvironmental factors – such as hypoxia and cells within GSC niches that sustain GSC status and modulate their phenotypes by upregulating GSC stress-signaling pathways and supplying GSCs with connective tissue components, growth factors and cytokines [Citation55,Citation56] – may establish a paracrine loop encouraging the U87 GSCs to recruit MSCs. Additionally, BLI presented unsatisfactory results in the non-invasive detection of the glioblastoma homing of IV-administered P-MSCs and was also unable to locate labelled cells without the reference of anatomical images. Since this represents a major limitation of imaging stem cell tracking, a more sensitive imaging system is needed.
Figure 3. Bioluminescent signal trafficking of P-MSCs in glioma-bearing mice. (A) Flowchart of experimental procedure. (B) Dorsal views of BLI of U87-bearing and U87 GSC-bearing mice that received luciferase-expressing P-MSCs (GFP-Fluc P-MSC) through IV injection. Representative images show the immediate (D0) and days 1, 3, 6 and 7 post-administration of P-MSCs. Signals were adjusted to the same color scale for each image. (C) Genomic DNA extracted from brain tissue of all mice survived on day 11 after P-MSCs inoculation was evaluated for luciferase gene and mouse CISD2 gene (as the DNA loading control) expression by PCR. DNA isolated from GFP-Fluc P-MSC was used as positive and negative controls for luciferase and CISD2, respectively.
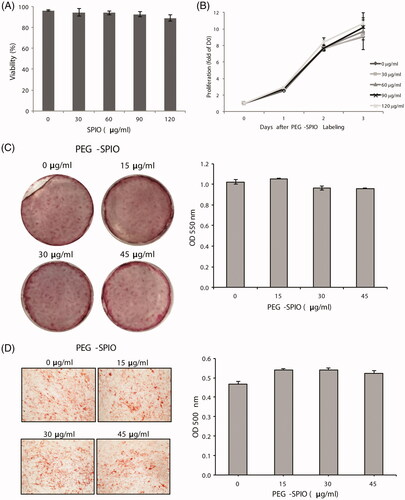
PEG–SPIO-labelled P-MSCs maintain MSC phenotypes
Cellular MRI is the primary imaging modality in most brain tumor cases. With unlimited depth and high spatial resolution (1–100 μm), the use of PEG-conjugated SPIO nanoparticles (PEG–SPIO) is a valuable tool for monitoring infiltration of stem cells [Citation57,Citation58]. We designed and synthesized PEG–SPIO as an MRI contrast agent for cellular imaging of P-MSCs (see section “Synthesis of PEG–SPIO and cell labeling” for details). We first characterized any influence of cellular labelling with PEG–SPIO on the viability and stemness of P-MSCs. Incubation of P-MSCs with PEG–SPIO caused no obvious cytotoxicity, even when the concentration of PEG–SPIO was 120 μg/ml (). After labelling, the P-MSCs retained an active proliferation capacity as unlabelled cells in vitro (). To verify whether PEG–SPIO labelling affected the ability of P-MSCs to differentiate towards multiple lineages, we assessed the adipogenic and osteogenic differentiation potency of P-MSCs in cell culture by supplementing with lineage-specific differentiation media. When P-MSCs were cultured in an osteogenic differentiation medium for 14 days, the cells differentiated into osteoblasts regardless of whether or not they were PEG–SPIO-labelled. This was demonstrated by Alizarin Red S staining (). Oil red O staining revealed that the PEG–SPIO-labelled P-MSCs also maintained adipocyte differentiation ability with a similar potency to that of the parental P-MSCs (). These results provide the in vitro evidence that PEG–SPIO nanoparticle labelling procedure has no adverse effects on cell growth or the stemness of P-MSCs.
Figure 4. Cytotoxicity and differentiation capabilities of PEG–SPIO-labelled P-MSCs. P-MSCs were incubated with the indicated concentration of PEG–SPIO for 15 min and then subjected to (A) trypan blue staining for cell viability assay, (B) WST-1 proliferation assay to determine cell growth rate, (C) Alizarin red S staining for osteogenic differentiation to detect bone mineralization and (D) Oil Red O staining for adipogenic differentiation to determine lipid droplet content. Assays were performed three times each. Data are presented as mean ± SD of one representative experiment; *P ≦ .05.
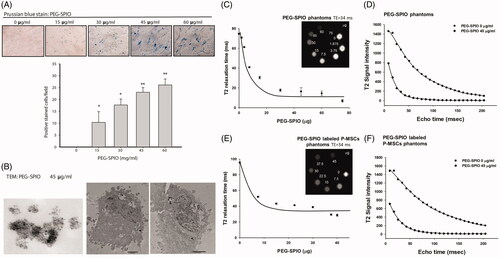
P-MSCs uptake PEG–SPIO and are obtained using MRI in vitro
Prior to in vivo tracking, we assessed the feasibility of MRI for imaging PEG–SPIO-labelled P-MSCs in vitro. Dose-dependent increased cellular uptake of PEG–SPIO by P-MSCs was first validated using Prussian staining (). Transmission electron microscopy (TEM) further confirmed that the intracellular homogeneous PEG–SPIO nanoparticles with diameters of less than 10 nm and the PEG–SPIO nanoparticles localized in the endosomal vesicles of P-MSCs, mostly near nuclei (). To determine the potency of the PEG–SPIO nanoparticles to be visualized using MRI, concentrations from 0 to 45 μg/mL PEG–SPIO were prepared as standard agarose (1%) phantoms for the MRI parameter setup. The T2 relaxation time was found to be markedly decreased with the escalation of PEG–SPIO dosage from as low as 7.5 μg/ml (50% reduction) (). The T2 signal fitting curve of 45 μg/mL shows that PEG–SPIO content rapidly decreased towards the background level (), suggesting that PEG–SPIO is a highly sensitive MRI contrast agent. After identifying visible PEG–SPIO concentrations in the phantom study, the cellular uptake of PEG–SPIO was detected by P-MSCs under the MRI scanner. By raising the PEG–SPIO dosage, the T2 relaxation time was reduced on the P-MSCs (). The amount of internalized PEG–SPIO was calculated up to 150 pg/cell based on the T2 relaxation curve made by PEG–SPIO standards. Through T2 fitting analysis, an obvious signal intensity decline was discovered in 45 μg/mL PEG–SPIO-labelled P-MSCs (). Based on the T2 fitting curve results, we are able to determine the most suitable T2 echo time for in vivo scanning. Relatively large signal differences between the PEG-SPIO-labelled and unlabelled P-MSCs were displayed at echo times of 0–50 ms, which is appropriate for in vivo trafficking.
Figure 5. In vitro validation of PEG–SPIO nanoparticles for MRI-based P-MSCs tracking. P-MSCs were incubated with the indicated concentration of PEG-SPIO nanoparticles for 15 min and then subjected to (A) Prussian blue staining for SPIO-positive cells and quantification of magnetic labelling efficiency using Image J software (*P ≦ .05 and **P ≦ .01), and (B) TEM to measure the size of PEG-SPIO nanoparticles (left, magnification = 150,000×) and determine cellular localization of nanoparticles (middle and right, magnification = 10,000×). T2 MR phantom images and quantified T2 relaxation time of (C) different amounts of PEG–SPIO nanoparticles alone and (E) its labelled P-MSCs under an 8.5-ms echo time. (D,F) Graph illustrates signal intensity (in arbitrary units) difference between 0 and 45 μg/mL PEG–SPIO phantoms alone and labelled P-MSCs.
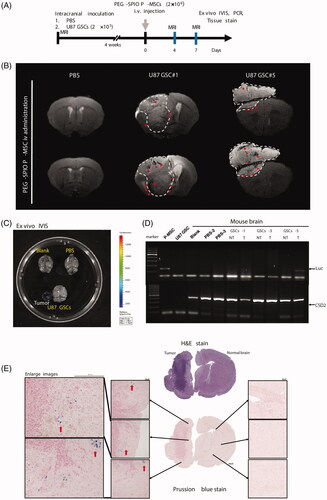
In vivo MRI effectively detects tumor infiltration of IV-administered PEG–SPIO-labelled P-MSCs in a U87 GSC animal model
We used MRI to validate whether IV-administered PEG–SPIO-labelled P-MSCs were detectable in GSCs-tumor bearing mice. The experimental procedure is outlined in . As demonstrated by T2-weighted MRI (T2 MRI) ( and Supplemental data, Figure S1), systemic administration of PEG–SPIO-labelled P-MSCs resulted in a clear T2 signal drop, with discontinuous amorphous dark curves at the margin of the tumor at 4 days after P-MSC transplantation. During follow-up examinations, the linear hypointense regions shown at day 4 developed into small and well-defined dark features located in the tumors at 7 days after transplantation, demonstrating the feasibility of in vivo MRI to image the temporal-spatial migration of systemically transplanted PEG-SPIO-labelled P-MSCs to glioma. In contrast, sham-control mice (PBS-group) infused with labelled P-MSCs did not develop hypointense signals with T2 MRI. The tropism effect of PEG–SPIO-labelled P-MSCs towards glioblastoma stem-like tumors was visible in the T2 MRI sequence in all survived mice. Nevertheless, although the sensitivity was relatively low in BLI and PCR for the detection of infiltrated P-MSCs in animal models, both methods demonstrated that the expression of luciferase, a transduced gene stably carried by P-MSCs, was found only in the extracted tumors showing negative enhancement of the T2 signal (). Moreover, a portion of mouse brains assessed for Prussian blue tissue staining showed no positive signal at the contralateral side; however, cells stained with Prussian blue were found clustered at the periphery of tumors, and some were scattered within tumors (). Collectively, these results confirmed the migratory potential of P-MSCs to intracranial U87 GSCs through IV administration. The IV transplanted P-MSCs were mainly trapped in the lungs, as demonstrated by BLI (), because they were larger than the diameter of the pulmonary capillaries. Nevertheless, the strong tumor-tracking properties of the P-MSCs still enabled a portion of cells to access the brain tumors and to be trafficked using the current form of PEG–SPIO cellular labelling agent with MRI.
Figure 6. In vivo validation of MRI for trafficking PEG–SPIO-labelled P-MSCs infiltration into mouse brain tumors. (A) Flowchart of experimental procedure. (B) T2 MRI of representative sham (PBS) and glioma-bearing (U87 GSC) mice on day 4 and day 7 after PEG–SPIO-labelled P-MSC injection. Significant hypointense spots (red arrowhead) developed at the tumor’s edge (white dashed line) or within the tumor area with the time in glioma-bearing mouse. (C) Ex vivo of BLI on mouse brains collected from normal (blank), sham (PBS) and glioma-bearing (U87 GSC) mice shows bioluminescence signals in the excised brain tumor of U87 GSC mouse #5. (D) PCR of luciferase gene in the genomic DNA isolated from individual mouse brain after 7 days of P-MSCs administration. Mouse CISD2 gene was used as the DNA loading control. (E) Prussian blue and H&E staining of entire brain sections harvested from U87 GSC-bearing mice. Red arrow indicates iron signal of P-MSCs.
Conclusions
This is the first report to demonstrate that hypoxia-preconditioned P-MSCs possess excellent BBB crossing and glioma tropism to GSCs, and the glioma trafficking behavior of P-MSCs labelled with PEG–SPIO nanoparticles can be non-invasively tracked through T2 MRI during systemic delivery in mouse models. Despite the use of surgery and improvements in radiotherapy and chemotherapy, the prognosis of patients with malignant glioma remains extremely poor. Therefore, we suggest that PEG–SPIO-labelled and hypoxia-preconditioned P-MSCs combined with cellular MRI modalities are appropriate for additional indication as systemic theranostics to maximize the therapeutic benefit for cerebral gliomas, especially targeting the aggressive tumor cells with stem-cell-like phenotypes.
Acknowledgments
The authors would like to acknowledge Misses Huei-Min Chen and Wen-Chie Wu for their excellent technical support on TEM and animal MRI at Taipei Medical University Core Facility.
Disclosure statement
The authors declared the following potential conflicts of interest: Dr. Chia-Ling Hsieh received research support from Meridigen Biotech Co. Ltd.; Drs. Willie Lin, Yogi Chang-Yo Hsuan and Yu-Chin Su are employees of Meridigen Biotech Co. Ltd.
Additional information
Funding
Related Research Data
References
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996.
- Lathia JD, Mack SC, Mulkearns-Hubert EE, et al. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217.
- Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;01414:105–111.
- Regina A, Demeule M, Laplante A, et al. Multidrug resistance in brain tumors: roles of the blood-brain barrier. Cancer Metastasis Rev. 2001;20:13–25.
- Woodworth GF, Dunn GP, Nance EA, et al. Emerging insights into barriers to effective brain tumor therapeutics. Front Oncol. 2014;4:126.
- Bao B, Ahmad A, Li Y, et al. Targeting CSCs within the tumor microenvironment for cancer therapy: a potential role of mesenchymal stem cells. Expert Opin Ther Targets. 2012;16:1041–1054.
- Bagó JR, Alfonso-Pecchio A, Okolie O, et al. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat Commun. 2016;7:10593.
- Zhang S, Xie R, Zhao T, et al. Neural stem cells preferentially migrate to glioma stem cells and reduce their stemness phenotypes. Int J Oncol. 2014;45:1989–1996.
- Theele DP, Schrimsher GW, Reier PJ. Comparison of the growth and fate of fetal spinal iso- and allografts in the adult rat injured spinal cord. Exp Neurol. 1996;142:128–143.
- Grisendi G, Bussolari R, Veronesi E, et al. Understanding tumor-stroma interplays for targeted therapies by armed mesenchymal stromal progenitors: the Mesenkillers. Am J Cancer Res. 2011;1:787–805.
- Kim SM, Lim JY, Park SI, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–9623.
- Saei Arezoumand K, Alizadeh E, Pilehvar-Soltanahmadi Y, et al. An overview on different strategies for the stemness maintenance of MSCs. Artif Cells Nanomed Biotechnol. 2017;45:1255–1271.
- Liu L, Eckert MA, Riazifar H, et al. From blood to the brain: can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013;2013:1.
- Gao F, Chiu SM, Motan DAL, et al. Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. 2016;7:e2062.
- Shi Y, Du L, Lin L, et al. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16:35–52.
- Kosztowski T, Zaidi HA, Quiñones-Hinojosa A. Applications of neural and mesenchymal stem cells in the treatment of gliomas. Expert Rev Anticancer Ther. 2009;9:597–612.
- Parolini O, Alviano F, Bagnara GP, et al. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells. 2008;26:300–311.
- Barlow S, Brooke G, Chatterjee K, et al. Comparison of human placenta- and bone marrow-derived multipotent mesenchymal stem cells. Stem Cells Dev. 2008;17:1095–1107.
- Brooke G, Tong H, Levesque JP, et al. Molecular trafficking mechanisms of multipotent mesenchymal stem cells derived from human bone marrow and placenta. Stem Cells Dev. 2008;17:929–940.
- Chambers DC, Enever D, Ilic N, et al. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19:1013–1018.
- Lublin FD, Bowen JD, Huddlestone J, et al. Human placenta-derived cells (PDA-001) for the treatment of adults with multiple sclerosis: a randomized, placebo-controlled, multiple-dose study. Mult Scler Relat Disord. 2014;3:696–704.
- Mayer L, Pandak WM, Melmed GY, et al. Safety and tolerability of human placenta-derived cells (PDA001) in treatment-resistant Crohn's disease: a phase 1 study. Inflamm Bowel Dis. 2013;19:754–760.
- Li X, Ling W, Pennisi A, et al. Human placenta-derived adherent cells prevent bone loss, stimulate bone formation, and suppress growth of multiple myeloma in bone. Stem Cells. 2011;29:263–273.
- Chen Q, Cheng P, Song N, et al. Antitumor activity of placenta-derived mesenchymal stem cells producing pigment epithelium-derived factor in a mouse melanoma model. Oncol Lett. 2012;4:413–418.
- Zheng L, Zhang D, Chen X, et al. Antitumor activities of human placenta-derived mesenchymal stem cells expressing endostatin on ovarian cancer. PLoS One. 2012;7:e39119.
- Frank JA, Anderson SA, Kalsih H, et al. Methods for magnetically labeling stem and other cells for detection by in vivo magnetic resonance imaging. Cytotherapy. 2004;6:621–625.
- Ariza de Schellenberger A, Kratz H, Farr TD, et al. Labeling of mesenchymal stem cells for MRI with single-cell sensitivity. Int J Nanomed. 2016;11:1517–1535.
- Naseri N, Ajorlou E, Asghari F, et al. An update on nanoparticle-based contrast agents in medical imaging. Artif Cells Nanomed Biotechnol. 2018;46:1111–1121.
- Jin R, Lin B, Li D, et al. Superparamagnetic iron oxide nanoparticles for MR imaging and therapy: design considerations and clinical applications. Curr Opin Pharmacol. 2014;18:18–27.
- Wang YX. Superparamagnetic iron oxide based MRI contrast agents: current status of clinical application. Quant Imaging Med Surg. 2011;1:35–40.
- Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17:484–499.
- Henning TD, Boddington S, Daldrup-Link HE. Labeling hESCs and hMSCs with iron oxide nanoparticles for non-invasive in vivo tracking with MR imaging. J Vis Exp. 2008;13:e685.
- Khurana A, Nejadnik H, Chapelin F, et al. Ferumoxytol: a new, clinically applicable label for stem-cell tracking in arthritic joints with MRI. Nanomedicine (Lond). 2013;8:1969–1983.
- Kitagawa F, Kubota K, Sueyoshi K, et al. One-step preparation of amino-PEG modified poly(methyl methacrylate) microchips for electrophoretic separation of biomolecules. J Pharm Biomed Anal. 2010;53:1272–1277.
- Chao YC, Su SK, Lin YW, et al. Synthesis and application of polyethylene glycol/vinyltriethoxy silane (PEG/VTES) copolymers. Afr J Biotechnol. 2011;10:12754–12761.
- Ling D, Hackett MJ, Hyeon T. Surface ligands in synthesis, modification, assembly and biomedical applications of nanoparticles. Nano Today. 2014;9:457–477.
- Shen R, Camargo PH, Xia Y, et al. Silane-based poly(ethylene glycol) as a primer for surface modification of nonhydrolytically synthesized nanoparticles using the Stober method. Langmuir. 2008;24:11189–11195.
- Kasten A, Gruttner C, Kuhn JP, et al. Comparative in vitro study on magnetic iron oxide nanoparticles for MRI tracking of adipose tissue-derived progenitor cells. PLoS One. 2014;9:e108055.
- Hu X, Wei L, Taylor TM, et al. Hypoxic preconditioning enhances bone marrow mesenchymal stem cell migration via Kv2.1 channel and FAK activation. Am J Physiol Cell Physiol. 2011;301:C362–C372.
- Pham K, Luo D, Liu C, et al. CCL5, CCR1 and CCR5 in murine glioblastoma: immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J Neuroimmunol. 2012;246:10–17.
- Pan Y, Smithson LJ, Ma Y, et al. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget. 2017;8:32977–32989.
- Ponte AL, Marais E, Gallay N, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737–1745.
- Ringe J, Strassburg S, Neumann K, et al. Towards in situ tissue repair: human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J Cell Biochem. 2007;101:135–146.
- Song CH, Honmou O, Furuoka H, et al. Identification of chemoattractive factors involved in the migration of bone marrow-derived mesenchymal stem cells to brain lesions caused by prions. J Virol. 2011;85:11069–11078.
- Deak E, Seifried E, Henschler R. Homing pathways of mesenchymal stromal cells (MSCs) and their role in clinical applications. Int Rev Immunol. 2010;29:514–529.
- Miao Z, Jin J, Chen L, et al. Isolation of mesenchymal stem cells from human placenta: comparison with human bone marrow mesenchymal stem cells. Cell Biol Int. 2006;30:681–687.
- Li G, Zhang XA, Wang H, et al. Comparative proteomic analysis of mesenchymal stem cells derived from human bone marrow, umbilical cord, and placenta: implication in the migration. Proteomics. 2009;9:20–30.
- Ho IA, Chan KY, Ng WH, et al. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009;27:1366–1375.
- Ries C, Egea V, Karow M, et al. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood. 2007;109:4055–4063.
- Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–3318.
- Hata N, Shinojima N, Gumin J, et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010;66:144–156.
- Thomas JG, Parker Kerrigan BC, Hossain A, et al. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J Neurosurg. 2018;128:287–295.
- Boltze J, Arnold A, Walczak P, et al. The dark side of the force - constraints and complications of cell therapies for stroke. Front Neurol. 2015;6:155.
- Cui LL, Kerkela E, Bakreen A, et al. The cerebral embolism evoked by intra-arterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cells Res Ther. 2015;6:11.
- Ye J, Wu D, Wu P, et al. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014;35:3945–3951.
- Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225–238.
- Arbab AS, Frank JA. Cellular MRI and its role in stem cell therapy. Regen Med. 2008;3:199–215.
- Wang Y-XJ, Quercy-Jouvet T, Wang H-H, et al. Efficacy and durability in direct labeling of mesenchymal stem cells using ultrasmall superparamagnetic iron oxide nanoparticles with organosilica, dextran, and PEG coatings. Materials. 2011;4:703–715.