
Abstract
Abnormal destruction of the components of the articular extracellular matrix (ECM) such as type II collagen and aggrecan caused by advanced glycation end products (AGEs) has been considered as one of the pathological characteristics of osteoarthritis (OA). Receptor-interacting protein 1 (RIP1), an important serine/threonine kinase, possesses a variety of biological functions including cell proliferation, survival and death. The physiological roles of RIP1 in OA have not been reported before. Here, we found that AGEs increased the expression of RIP1 in human chondrosarcoma cell line SW1353 cells. Importantly, we found that antagonism of RIP1 using its specific inhibitor necrostatin-1 (Nec-1) ameliorated AGE-induced degradation of type II collagen and aggrecan in SW1353 cells. We also found that treatment with Nec-1 reduced the expression of MMP-3 and MMP-13 but restored the expression of Tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. Also, our results indicate that Nec-1 inhibited AGE-induced expression of ADAMTS-4 and ADAMTS-5. Mechanistically, we found that Nec-1 treatment inhibited the activation of JNK and the transcriptional factor AP-1 by reducing the expressions of c-Fos and c-Jun, the two main components of AP-1. Additionally, we found that Nec-1 treatment abolished AGE-induced activation of the transcriptional factor NF-κB by suppressing the nuclear translocation of p65. These findings suggest that RIP1 might be an important therapeutic target of OA.
Introduction
Osteoarthritis (OA) is one of the most common debilitating diseases among elderly people and can be attributed to the aging process, obesity, trauma, genetic factors and endocrine factors; however, the exact mechanisms driving the progression of this disease remain unclear. In the pathological processes behind the development of OA, excessive destruction of the articular extracellular matrix (ECM) plays a pivotal role [Citation1,Citation2]. Type II collagen acts as the structural support of the ECM in human articular cartilage. Joint damage resulting from degradation of type II collagen by MMPs, such as MMP-3 and -13, is a crucial event in the progression of OA [Citation2]. Of these, MMP-3 acts as a stromelysin, thereby activating MMP-13, among others. Tissue inhibitor of metalloproteinases (TIMPs), such as TIMP-1 and -2, strongly antagonize MMP activity, and therefore maintaining a proper balance between TIMPs and MMPs is crucial for the prevention of OA development [Citation3]. Additionally, aggrecanases such as ADAMT-4 and -5 have been shown to induce degradation of the ECM by causing cleavage of proteoglycans and aggrecans within the matrix. Advanced glycation end products (AGEs) result from non-enzymatic protein glycation and due to their resilience against degradation; accumulate in joint tissues [Citation2]. Receptor of AGEs (RAGE) is expressed in articular chondrocytes and synovial tissue macrophages in rheumatoid arthritis (RA), and has been found to stimulate chondrocytes and synoviocytes in patients with OA, thereby inducing an increase in the production of MMP-3, MMP-13 and TNF-α by OA chondrocytes. TNF-α also induces further release of MMPs, thereby exacerbating and sustaining MMP-induced degradation of type II collagen in the ECM [Citation4]. The involvement of intracellular signalling pathways in the degradation of ECM is complicated. So far, stress factors such as AGE-induced activation of mitogen-activated protein kinases (MAPK) and JNK/c-Fos/AP-1 have been reported to mediate the expression of MMPs in chondrocytes. Additionally, the transcriptional factor NF-κB plays a pivotal role in regulating the expression of MMPs and ADAMTs [Citation5]. A previous study demonstrated that expression of MMP-13 induced by TNF-α could be down-regulated via inhibition of the AP-1 and NF-κB transcription factors in primary human chondrocytes and SW1353 cells, thereby suggesting that these transcription factors mediate the induction of MMP-13 by TNF-α [Citation6].
Receptor-interacting protein 1 (RIP1) kinase is well recognized as a key upstream regulator of NF-κB activation induced by TNF-α as well as an activator of apoptosis and necroptosis, a pro-inflammatory type of programmed cell death. RIP1 modulates these pathways through ubiquitylation, deubiquitylation and ubiquitin receptor interactions and is considered an important target for the treatment of both acute and chronic diseases [Citation7]. RIP1 has been found to activate NF-κB, Akt and inhibit p53 activation in glioblastoma cells while overexpression of RIP1 in glioblastoma cells is associated with poorer prognosis. Additionally, it has been suggested that RIP1 plays a role in promoting tumour survival by protecting DNA-damaged cells against cytotoxicity associated with excessive production of reactive oxygen species (ROS) [Citation8]. Necrostatin-1 (Nec-1), a specific inhibitor of RIP1, has been demonstrated to exert anti-apoptotic and anti-necroptotic effects in a variety of diseases including ischemia/reperfusion injury and brain injury following intracerebral haemorrhage [Citation9,Citation10]. This small molecule inhibitor of RIP1 Ser/Thr kinase activity was initially employed as an inhibitor of necroptosis, which is dependent on RIP1 kinase. However, the biological functions of RIP1 and the pharmacological roles of Nec-1 in OA and ECM metabolism in human chondrocytes remain unknown. In this study, we used Nec-1 to treat human chondrosarcoma SW1353 cells to investigate the role of RIP1 as a potential regulator of ECM degradation in articular cartilage.
Materials and methods
Preparation of AGEs
Methylglyoxal-modified albumin was used to prepare AGEs. Ten mg/mL BSA was incubated with 50 mM methylglyoxal containing sodium azide (0.1%) and phenylmethylsulfonyl fluoride (PMSF) (1 mM) at 37 °C for 7 d, followed by a gentle dialysis in phosphate-buffered saline (PBS) overnight.
Cell culture and treatment
Human chondrosarcoma SW1353 cell line was purchased from ATCC. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, glutamine, and 0.1% penicillin and streptomycin. Cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h.
Real-time polymerase chain reaction
Total intracellular RNA was extracted with Qiazol reagent (Qiagen, USA). DNase I was used to remove DNA contamination. cDNA was prepared using 2 μg of total intracellular RNA with an iScript reverse transcription kit (Bio-Rad, USA). Real-time polymerase chain reaction (PCR) experiments were performed with template cDNA samples on the ABI 7500 real-time PCR system. Experimental results were analysed by normalizing to GAPDH using the 2–△△Ct method.
Nuclear protein extraction
Nuclear proteins were extracted from SW1353 cells using a commercial protein extraction kit (Thermo Scientific, USA). After the necessary treatment, SW1353 cells were lysed in cell lysis buffer. Samples were then vortexed vigorously for 10 s, immediately followed by centrifugation for 30 s at 12,000 × g. Pellets were collected and resuspended in cell lysis buffer. After gentle centrifugation at 20,000 × g for 15 min, the supernatant was collected and used for western blot analysis. The nuclear specific protein lamin B was used as a positive control for nuclear extractions.
Protein isolation and Western blots
Protein extractions were prepared using cell lysis buffer containing inhibitor cocktail. Protein concentrations were measured using the bicinchoninic acid (BCA) method. Equal amounts of intracellular protein (20 μg) were separated by 8%–12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PDVF) membrane (Amersham, USA). The membrane was then blocked with 5% non-fat milk and incubated with primary antibodies overnight at 4 °C. After washing three times with tris-buffered saline and Tween 20 (TBST), membranes were incubated with horse radish peroxide (HRP)-conjugated secondary antibodies. Immunoblot signals were visualized using enhanced chemiluminescent (ECL) substrate.
Statistical analysis
Experimental results were analysed using SPSS version 19 software (SPSS, Germany). Data are presented as means ± standard deviation. Statistical differences were calculated using univariate analysis of variance (ANOVA). p < .05 was considered statistically significant.
Results
Excessive accumulation of AGEs has been reported to be involved in the pathological development of OA. Firstly, we investigated the effects of AGEs on the expressions of RIP1 in human chondrosarcoma cell line SW1353 cells. Here, we found that the expression of RIP1 was increased in response to treatment with AGEs in a dose-dependent manner at the concentrations of 50, 100 and 200 μg/mL at both the mRNA () and protein levels ().
Figure 1. AGEs increased the expression of RIP1 in human chondrosarcoma SW1353 cells. SW1353 cells were treated with 50, 100 and 200 μg/mL AGEs for 24 h. (A) Real-time PCR analysis of RIP1. (B) Western blot analysis of RIP1 (*, #, $, p < .01 vs. previous column group).

To investigate the effects of RIP1 on AGEs-induced degradation of type II collagen and aggrecan, human SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1, a specific inhibitor of RIP1, at concentrations of 50 and 100 µM for 24 h. The western blot results shown in indicate that treatment with 100 μg/mL AGEs caused significant degradation of type II collagen and aggrecan, which could be preserved by treatment with Nec-1 in a dose-dependent manner ( and (B)).
Figure 2. The specific RIP1 inhibitor Nec-1 ameliorated AGE-induced degradation of type II collagen and aggrecan. Human chondrosarcoma SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Representative images of western blot analysis of type II collagen and aggrecan. (B) Quantification of type II collagen and aggrecan (*, #, $, p < .01 vs. previous column group).
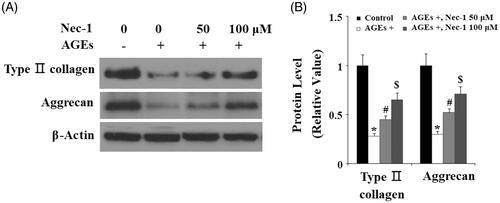
The degradation of type II collagen is mainly mediated by MMP-3 and MMP-13. Here, the effects of Nec-1 on MMP expression were investigated. The real-time PCR analysis in indicated that treatment with AGEs significantly increased the expression of MMP-3 and MMP-13 at the gene level. Interestingly, the presence of Nec-1 significantly reduced the expression of MMP-3 and MMP-13 in a dose-dependent manner. These results were verified by western blot analysis at the protein level (). Tissue inhibitor of metalloproteinase-1 and -2 (TIMP-1 and TIMP-2) are important inhibitors of MMPs, including MMP-3 and MMP-13. Here, we found that AGEs remarkably reduced the expression of both TIMP-1 and TIMP-2, which were ameliorated by treatment with Nec-1 in a dose-dependent manner at both the mRNA level () and the protein level ().
Figure 3. The specific RIP1 inhibitor Nec-1 ameliorated AGE-induced expression of MMP-3 and MMP-13. Human chondrosarcoma SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Real-time PCR analysis of MMP-3 and MMP-13. (B) Western blot analysis of MMP-3 and MMP-13 (*, #, $, p < .01 vs. previous column group).
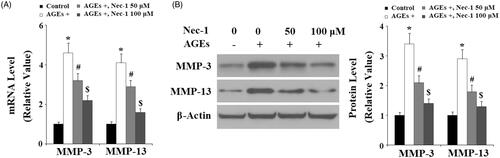
Figure 4. The specific RIP1 inhibitor Nec-1 ameliorated AGE-induced reduction of TIMP-1 and TIMP-2. Human chondrosarcoma SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Real-time PCR analysis of TIMP-1 and TIMP-2. (B) Western blot analysis of TIMP-1 and TIMP-2 (*, #, $, p < .01 vs. previous column group).
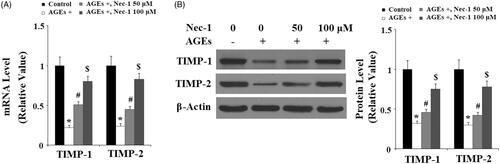
The disintegrin and metalloproteinase with thrombospondin motifs-4 and -5 (ADAMTS-4, -5) are responsible for aggrecan turnover in articular cartilage. Here, we found that AGEs significantly promoted the expressions of both ADAMTS-4 and ADAMTS-5, which were suppressed by Nec-1 at both the mRNA level () and the protein level () in a dose-dependent manner.
Figure 5. The specific RIP1 inhibitor Nec-1 ameliorated AGE-induced expression of ADAMTS-4 and ADAMTS-5. Human chondrosarcoma SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Real-time PCR analysis of ADAMTS-4 and ADAMTS-5. (B) Western blot analysis of ADAMTS-4 and ADAMTS-5 (*, #, $, p < .01 vs. previous column group).
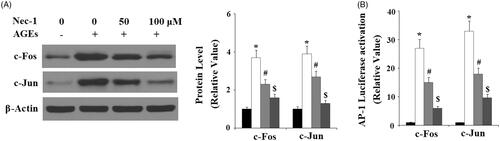
To disclose the molecular mechanisms of Nec-1 in ECM degradation, we investigated the JNK pathway. Here, we found that the presence of Nec-1 significantly ameliorated AGE-induced phosphorylation of JNK () in a dose-dependent manner. However, the total level of JNK remained consistent. Phosphorylation of JNK can promote activation of the transcriptional factor AP-1. Here, we investigated the effects of Nec-1 on AP-1. C-fos and c-Jun are the two main subunits of AP-1. Here, we found that AGE-induced expression of c-fos and c-Jun were significantly inhibited by Nec-1 treatment in a dose-dependent manner (). We transfected the c-Jun/c-Fos containing promoter AP-1 into SW1353 cells. Importantly, we found that AGEs significantly increased the luciferase activity of AP-1, which was suppressed by Nec-1 treatment (). These findings implicate that the JNK/AP-1 signalling pathway played an important role in the effects of Nec-1.
Figure 6. Nec-1 suppressed AGE-induced activation of JNK. Human chondrosarcoma SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 1 h. (A) Phosphorylated and total levels of JNK. (B) Quantification of phosphorylated JNK (*, #, $, p < .01 vs. previous column group).
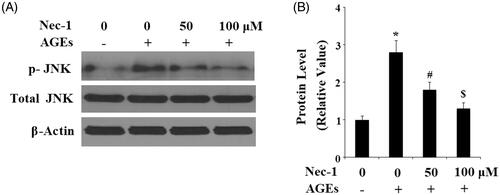
Figure 7. Nec-1 suppressed AGE-induced activation of transcription factor AP-1. Human chondrosarcoma cell line SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Western blot analysis of c-Fos and c-Jun. (B) Luciferase reporter assay demonstrated that Nec-1 treatment suppressed AP-1 activation in a dose-dependent manner (*, #, $, p < .01 vs. previous column group).
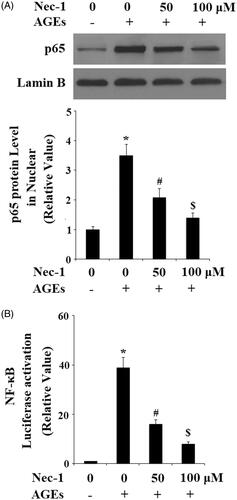
The NF-κB signalling pathway has been reported to be involved in the degradation of ECM in human chondrocytes. Here, we found that treatment with AGEs significantly increased the nuclear level of p65, which was prevented by treatment with Nec-1 in a dose-dependent manner (). Notably, the presence of Nec-1 reduced the luciferase activity of the NF-κB promoter ().
Figure 8. Nec-1 suppressed AGE-induced activation of NF-κB. Human chondrosarcoma cell line SW1353 cells were treated with 100 μg/mL AGEs in the presence or absence of Nec-1 (50 and 100 μM) for 24 h. (A) Nuclear level of p65. (B) Luciferase reporter assay demonstrated that Nec-1 treatment suppressed NF-κB activation in a dose-dependent manner (*, #, $, p < .01 vs. previous column group).
Discussion
One of the main hallmarks of OA, degradation of the articular ECM results in the loss of structural integrity and subsequent destruction of the joint, creates a great impact on mobility and quality of life. Many factors can contribute to the development and progression of this disease. While joint trauma from mechanical injury, physical activity or obesity can lead to the development of OA in younger people; it is most common among the elderly [Citation11,Citation12]. As the age of the global population increases, it becomes more and more important to elucidate the mechanisms behind age-related diseases such as OA and to develop both preventative and restorative therapies.
Beyond physical joint damage caused by the long-term mechanical stress and increased likelihood of obesity associated with advanced age, there are numerous molecular processes that contribute to the development and progression of OA. Glycation, also known as the Maillard reaction, is a process that takes place between amino groups of biomacromolecule and reducing sugars and results in the production of ROS and AGEs. Excess ROS leads to enhanced glycation and increased accumulation of AGEs. Accumulation of AGEs accompanies the natural process of aging and has been associated with build-up of plaques in Alzheimer’s and Parkinson’s diseases, complications associated with diabetes, and the development and progression of OA [Citation13,Citation14]. A recent in vivo study in which rabbits were injected with exogenous AGEs revealed that exposure of chondrocytes to AGEs resulted in increased production of ROS and release of cytochrome c, and significantly decreased cell viability, mitochondrial membrane potential and production of ATP in a time- and dose-dependent manner [Citation15]. In this study, we aimed to investigate the molecular mechanism through which AGEs exert their toxic effects on chondrocytes using the RIP1-specific inhibitor Nec-1.
While RIP1-deficient mouse models have proven to be unviable, a study using RIP1 kinase-dead (Ripk1K45A) mice revealed that the kinase activity of RIP1 is not required for maintaining viability and homeostasis, and that silencing of RIP1 kinase provided a protective effect against necroptotic stimuli both in vitro and in vivo [Citation16]. Recently, RIP1 was identified as playing a central role in insulin resistance, obesity and atherosclerosis in a study using atherosclerotic and diet-induced obesity RIP1-knockdown mouse models [Citation17]. Here, we found that RIP1 expression was increased in response to treatment with AGEs in a dose-dependent manner () while inhibition of RIP1 using Nec-1 led to a significant decrease in AGE-induced degradation of type II collagen and aggrecan in SW1353 chondrocytes (. Numerous studies have identified MMP-3 and MMP-13 as playing a major role in mediating degradation of ECM type II collagen in OA joints [Citation18,Citation19]. Consistent with the results of this study, AGEs have been found to increase expression of MMP-3 and -13 [Citation20,Citation21]. The results shown in indicate that treatment with Nec-1 ameliorated the increased expression of MMP-3 and -13 induced by AGEs. TIMP-1 and -2 antagonize the activities of MMPs and therefore maintaining the correct ratio of MMPs to TIMPs is crucial for preventing the progression of OA [Citation3,Citation22,Citation23]. As revealed in this study, expression of TIMP-1 and -2 is significantly reduced upon exposure of chondrocytes to AGEs. This increase in expression of MMP-3 and -13 and simultaneous decrease in expression of TIMP-1 and -2 induced by AGEs leads to a significant imbalance in the ratio of MMPs to TIMPs, thereby exacerbating the pathological progression of OA. As shown in , treatment with Nec-1 also ameliorated the inhibitory effect of AGEs on the expression of TIMP-1 and -2. These results implicate the essential role of RIP1 in regulating degradation of the ECM in human chondrocytes.
Along with the destruction of type II collagen, degradation of aggrecan by aggrecanases such as ADAMTS-4 and -5 has been associated with destruction of the ECM in OA joints. Of these, ADAMTS-4 has been considered as the major enzyme responsible for cleavage of the Glu373–Ala374 bond in aggrecan in humans [Citation24]. However, previous studies have also shown that deletion of ADAMTS-5 can prevent cartilage degradation in OA mouse models [Citation25]. Here, we investigated the effect of Nec-1 treatment on the expression of ADAMTS-4 and -5 and found that while exposure to AGEs increased expression of these aggrecanases, treatment with Nec-1 suppressed the expression of ADAMTS-4 and -5 in a dose-dependent manner (.
A study by Thalhamer et al. revealed that MAPKs such as ERK, p38 MAPK and JNK play an important role in regulating the expression of MMPs [Citation26]. Increased phosphorylation of JNK induced by AGEs can promote activation of the transcriptional factor AP-1. Both AP-1 and NF-κB have been shown to mediate a wide spectrum of inflammatory reactions including chondrocyte dysfunction in articular cartilage in OA [Citation27]. The inhibitory effects of Nec-1 on AP-1 and NF-κB activation suggest that RIP1 may be a potential therapeutic target for the treatment of OA.
In summary, this study determined that blockage of RIP1 using Nec-1 has the potential for use in the amelioration of type II collagen and aggrecan degradation. This beneficial effect of Nec-1 was correlated to inhibition of MMPs and ADAMTSs via regulation of the AP-1 and NF-κB pathway. These observations suggest that inhibition of RIP1 using Nec-1 may have the potential to protect chondrocytes against AGEs-mediated cartilage matrix destruction in arthritis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Hu G, Zhao X, Wang C, et al. MicroRNA-145 attenuates TNF-α-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017;8:3140.
- Huang CY, Lai KY, Hung LF, et al. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathways in porcine chondrocytes. Rheumatology 2011;50:1379–1389.
- Qu H, Li J, Wu L, et al. Trichostatin A increases the TIMP-1/MMP ratio to protect against osteoarthritis in an animal model of the disease. Mol Med Rep. 2016;14:2423–2430.
- Nah SS, Choi IY, Yoo B, et al. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-α in human osteoarthritic chondrocytes. FEBS Lett. 2007;581:1928–1932.
- Lim H, Kim HP. Matrix metalloproteinase-13 expression in IL-1β-treated chondrocytes by activation of the p38 MAPK/c-Fos/AP-1 and JAK/STAT pathways. Arch Pharm Res. 2011;34:109.
- Liacini A, Sylvester J, Li WQ, et al. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp Cell Res. 2003;288:208–217.
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–736.
- Wang Q, Chen W, Xu X, et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis. 2013;34:2119–2128.
- Koudstaal S, Oerlemans MI, Van der Spoel TI, et al. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur J Clin Invest. 2015;45:150–159.
- Su X, Wang H, Kang D, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem Res. 2015;40:643.
- Singh M, Sharma AR, Ummat A. An observational study-correlation of osteoarthritis knee with BMI, age and gender in a tertiary care hospital in Mullana. Ambala. Int J Sci Res. 2018;7:72–73.
- Hootman JM, Macera CA, Helmick CG, et al. Influence of physical activity-related joint stress on the risk of self-reported hip/knee osteoarthritis: a new method to quantify physical activity. Prev Med. 2003;36:636–644.
- DeGroot J, Verzijl N, Wenting V, et al. Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis. Arthritis Rheum. 2004;50:1207–1215.
- Bohlooli M, Mansour G, Khajeh M, et al. Acetoacetate promotes the formation of fluorescent advanced glycation end products (AGEs). J Biomol Struct Dynam. 2016;34:1–2666.
- Yang Q, Guo S, Wang S, et al. Advanced glycation end products-induced chondrocyte apoptosis through mitochondrial dysfunction in cultured rabbit chondrocyte. Fundam Clin Pharmacol. 2015;29:54–61.
- Berger SB, Kasparcova V, Hoffman S, et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480.
- Karunakaran D, Nguyen MA, Geoffrion M, et al. Therapeutic inhibition of RIP1 improves metabolic dysfunction and inhibits atherosclerosis in mouse models of cardiometabolic diseases. FASEB J. 2018;32(supplement):38.1–38.1.
- Freije JM, Diez-Itza I, Balbin M, et al. Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem. 1994;269:16766–16773.
- Yamamoto K, Okano H, Miyagawa W, et al. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016;56:57–73.
- Huang W, Ao P, Li J, et al. Autophagy protects advanced glycation end product-induced apoptosis and expression of MMP-3 and MMP-13 in rat chondrocytes. BioMed Res Int. 2017;2017:6341919.
- Peeters SA, Engelen L, Buijs J, et al. Associations between advanced glycation endproducts and matrix metalloproteinases and its inhibitor in individuals with type 1 diabetes. J Diab Complic. 2018;32:325–329.
- Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727.
- Johnston P, Larson D, Clark IM, et al. Metalloproteinase gene expression correlates with clinical outcome in Dupuytren's disease. J Hand Surg Am. 2008;33:1160–1167.
- Verma P, Dalal K, Chopra M. Pharmacophore development and screening for discovery of potential inhibitors of ADAMTS-4 for osteoarthritis therapy. J Mol Model. 2016;22:178.
- Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648.
- Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation, Rheumatology, 2008;47(4):409–414.
- Saito T, Tanaka S. Molecular mechanisms underlying osteoarthritis development: notch and NF-κB. Arthritis Res Ther. 2017;19:94.