
Abstract
Background
Although dexmedetomidine (Dex) has a significant neuroprotective effect in various nerve-damage models, the exact mechanism of which Dex protects cells from oxidative damage is not fully clear. This article recommended the protective effect of Dex on oxidative damage in PC12 cells.
Methods
The PC12 cells were incubated by hydrogen peroxide (H2O2) for 24 h and pre-treated by Dex for 30 min. Cell viability, apoptosis, HIF-1α expression and ROS level were detected by CCK-8, apoptosis assay, Western blot and ROS assay, respectively. The miR-199a expression was tested by qRT-PCR. Targeting relationship between miR-199a and HIF-1α was performed by dual luciferase activity assay. The activation of PI3K/AKT/mTOR and Wnt/β-catenin pathways was tested by western blot.
Results
Dex attenuated H2O2-induced oxidative damage, including the decline of cell viability, the raise of apoptosis and the generation of ROS in PC12 cells by down-regulating miR-199a expression. Moreover, Dex up-regulated HIF-1α expression via decreasing miR-199a level in PC12 cells and miR-199a targeted the 3′-UTR of HIF-1α. In addition, Dex activated PI3K/AKT/mTOR and Wnt/β-catenin pathways by declining miR-199a level.
Conclusions
This article illustrated the protective effect of Dex on oxidative damage in PC12 cells. Furthermore, Dex prevented PC12 cells from oxidative injury through the regulation of miR-199a/HIF-1α.
Introduction
It is well known that the neurodegenerative disease often occurs in middle-aged people in recent years, and the annual incidence is constantly increasing. The disease’s features are pathological changes in specific brain regions and denatured death of different neurons. The studies showed that oxidative damage and cell apoptosis played a significant role in the development of neurodegenerative diseases [Citation1]. By the same token, a study showed that reactive oxygen species (ROS) was able to damage neurons [Citation2]. Thus, it is fairly necessary to find a new drug to protect nerve cells from oxidative damage and thus act as a neuroprotective agent.
Dexmedetomidine (Dex) is a selective alpha 2 adrenergic receptor (α2AR) agonist with sedative, anxiolytic, antisympathetic and analgesic effects for intraoperative and postoperative management [Citation3]. It has been widely used in perioperative anaesthesia and intensive care unit. In clinical researches, not only the protective effects of Dex are closely related to activate α2AR signalling pathway, but also other signalling pathways (such as PI3K/Akt) are involved in the protective impacts of Dex [Citation4,Citation5]. There was also increasing evidence illustrated that Dex had significant neuroprotective effects in a variety of neural injury models. For example, Dex could relieve neuroinflammation induced by LPS [Citation6]. The study showed that Dex could protect neurons and astrocytes against β-amyloid (Aβ) [Citation7]. Although countless studies have reported the neuroprotection of Dex, it is seldom informed that Dex plays the role of neuroprotection through anti-oxidant damage.
There have been studies reported that microRNAs (miRNAs) play a crucial significance in the improvement of neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) [Citation8–10]. For example, the activation of miR-3557/324 could regulate the CaMKs signalling pathway to decrease the incidence of PD [Citation11]. The decline of miR-101a level accelerated the progress of AD [Citation12]. Moreover, a study showed that the decline of miR-199a-5p, as well as miR-199a-3p levels, was involved in PD [Citation13]. In addition, only a few studies have illustrated miR-199a is closely related to hypoxia-adaptation [Citation14–17]. So, we speculated if the level of miR-199a expression participated in oxidative damage in neurodegenerative diseases.
The PC12 cells are derived from Rattus norvegicus adrenal pheochromocytoma (a tumour of the sympathetic nervous system) [Citation18]. PC12 cell line is frequently used as a model system for neurobiological and neurochemical studies [Citation19,Citation20]. To sum up, the protective effects of Dex on oxidative damage were studied in PC12 cells. This article brought evidence to explain if Dex protected PC12 cells against oxidative injury via the regulation of miR-199a/HIF-1α.
Materials and methods
Cell culture and treatment
The PC12 cells were received from the Kunming Institute of Zoology (Kunming, China). The cells were brood in DMEM medium (Thermo Fisher, Waltham, MA) along with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (both from PAN Biotech, Aidenbach, Germany). Cells were seeded (1 × 104 cells/mL). It was maintained in a chamber (Thermo Fisher, Waltham, MA) at 37 °C embracing 5% CO2.
The hydrogen peroxide (H2O2) solution was gained from Sigma-Aldrich (cat. number: 323381, St. Louis, MO). PC12 cells were incubated by various concentrations of H2O2 (0, 12.5, 50, 100 and 200 µM) for 24 h. The Dex was purchased from Hengrui Medicine Co., Ltd (cat. number: 15030932, Lianyungang, China) and dissolved in DMEM medium. After the PC12 cells were pre-treated by Dex (1 nM) for 30 min, cells were incubated by 50 µM H2O2 for 24 h.
Cell transfection
The respective negative control (NC mimic) and miR-199a mimic were compounded by GenePharma Co. (Shanghai, China) and the transfection concentration of NC mimic and miR-199a mimic was 100 nM and 50 nM, respectively. The sequence of miR-199a mimic was 5′-CCCAGUGUUCAGACUACCUGUUC-3′. Transfection was conducted by adopting lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). The time of transfection was set as 48 h, in line with manufacturers’ contract.
Cell counting kit-8 (CCK-8) assay
Cells were celled as a density of 5000 cells per well in 96-well plates. After cells were treated by drugs, the CCK-8 solution (Dojindo Molecular Technologies, Gaithersburg, MD) was picked up into every well. After CCK-8 solution cultured cells for 1 h, the absorbance was computed by operating a Microplate Reader (Bio-Rad, Hercules, CA, USA) at a wavelength of 450 nm.
Apoptosis assay
Cell apoptosis was completed by collecting propidium iodide (PI) and fluorescein isothiocyanate (FITC)-conjugated Annexin V staining. After all the cells were treated with distinctive drugs, cells were washed by using phosphate-buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO). Then cells were coloured in 100 µL PI/FITC-Annexin V (Beijing Biosea Biotechnology, Beijing, China). Flow cytometry test was performed in agreement with the instruction of FACS can (Beckman Coulter, Fullerton, CA). FITC-positive and PI-negative cells were counted as apoptotic cells. All data were counted by FlowJo software (Treestar, San Carlos, CA).
ROS assay
PC12 cells were mixed and celled in six-well plates. After cells were incubated by different drugs, cells washed by PBS then were brood by medium without serum incorporating 10 μM DCFH-DA (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37°C. Subsequently, cells washed were suspended with 500 μL PBS again. Finally, the fluorescent degrees were calculated by applying a flow cytometer (Beckman Coulter, Fullerton, CA, USA). The excitation was set as 488 nm and the emission was set as 521 nm, respectively.
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
The entire RNA was isolated by utilizing Trizol reagent (Life Technologies Corporation, Carlsbad, CA). Single-step cDNA synthesis and miRNA quantitation were performed by using Mir-X™ miRNA First Strand Synthesis Kit and Mir-X™ miRNA qRT-PCR TB Green™ Kit (Takara, Dalian, China), respectively. A specific forward primer of miR-199a (5′ACACTCCAGCTGGGCCCAGTGTTCAGACTAC-3′) and the reverse primer of miR-199a (5′-TGGTGTCGTGGAGTCG-3′) were used in this experiment. U6 (the forward primer: 5′-CAAATTCGTGAAGCGTT-3′ and the reverse primer: 5′-TGGTGTCGTGGAGTCG-3′) was thought as the control of miRNA. The data were analyzed by using 2–ΔΔCt method.
Dual luciferase activity assay
The 3′-UTR regain of HIF-1α was amplified by PCR and placed into pGL3 reporter vector to form a reporter plasmid comprising the wide type of HIF-1α 3′-UTR (HIF-1α-wt). The anticipated combining site of the 3′-UTR of HIF-1α which can bind with miR-199a was mutated and inserted to form a control plasmid of HIF-1α-mut. The plasmids were transfected together with miR-199a mimic or NC mimic into 293T cell (all from Promega, Madison, WI). Reporter assays were accomplished by performing the dual-luciferase assay system (Promega, Madison, WI) in consonance with the directive.
Western blot
Proteins in PC12 cells were obtained through collecting a protein extraction kit (Sigma-Aldrich, St. Louis, MO). The concentration of those was counted by using BCA™ Protein Assay Kit (Pierce, Appleton, WI). A Bio-Rad Bis-Tris Gel system was used to separate proteins in consonance with manufacturer’s guidance. The membranes (Millipore, Billerica, MA, USA) were covered by 5% bovine serum albumin (BSA) (from Sigma-Aldrich, St. Louis, MO, USA). The experimental involved primary antibodies against Bcl-2 (ab185002, Abcam, Cambridge, UK), Bax (ab32503), pro-caspase-3 (ab238440), cleaved-caspase-3 (ab2302), HIF-1α (ab1), t-PI3K (ab86714), p-PI3K (ab182651), p-AKT (ab38449), t-AKT (ab8805), t-mTOR (ab2732), β-catenin (ab32572), Wnt3a (ab219412), β-actin (ab8227) and p-mTOR (#5536, Cell Signaling Technology, Beverly, MA, USA). Two kinds of second antibodies (goat anti-rabbit (ab6940, Abcam, Cambridge, UK) and goat anti-mouse (ab97035)) were used. Positive bands were developed by BeyoECL Moon (Beyotime, Shanghai, China). Different protein was estimated by Image Lab™ Software (Bio-Rad, Shanghai, China).
Statistical analysis
Three experiments were completed to repeat every result. All data are shown as mean + SD. We adopted Graphpad statistical software 6.0 (Graphpad Prism, Chicago, IL) to analysis our data. Comparison between the two groups was tested by t test, and a comparison between three or more was adopted by one-way analysis of variance (ANOVA). We made a conclusion that p < .05 was regarded as a meaningful result.
Results
Dex attenuated H2O2-evoked oxidative damage in PC12 cells
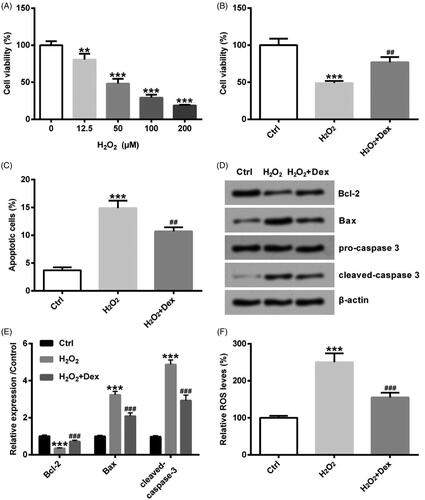
PC12 cells were incubated by distinctive concentrations of H2O2 (0, 12.5, 50, 100 and 200 µM) for 24 h. As described in , cell viability showed the declined trends with the concentration of H2O2 increasing. When the concentration of H2O2 was 12.5 µM, cell viability was started to be lowered (p < .05). As cell viability was about 50% when the concentration of H2O2 was 50 µM, 50 µM was used as following experimental concentration. After pre-treatment by Dex (1 nM) for 30 min, PC12 cells were incubated by 50 µM H2O2 for 24 h. As displayed in , co-treatment of cells with Dex and H2O2 significantly increased cell viability as compared to treatment with H2O2 alone (p < .05). Rate of cell apoptosis was markedly raised in the H2O2 group, and the rising rate of cell apoptosis induced by H2O2 was partly reversed by Dex (both p < .05, ). The Bcl-2 protein level was strikingly down-regulated, but the levels of Bax, as well as cleaved-caspase-3, were obviously raised by H2O2 (all p < .05, ). The above three proteins’ changes induced by H2O2 were reversed by Dex (all p < .05, ). The ROS level was significantly increased by H2O2, but Dex decreased ROS levels (both p < .05, ). Above all results explained that Dex attenuated H2O2-evoked oxidative injury in PC12 cells.
Figure 1. Dex attenuated H2O2-evoked oxidative damage in PC12 cells. Cells were incubated by different concentrations of H2O2 (0, 12.5, 50, 100 and 200 µM) for 24 h. (A) Cell viability was computed by CCK-8 assay. After pre-treatment by Dex (1 nM) for 30 min, PC12 cells were incubated by 50 µM H2O2 for 24 h. (B) Cell viability was tested by CCK-8, (C) the rate of cell apoptosis was counted by apoptosis assay, (D, E) the levels of Bcl-2, Bax and cleaved-caspase 3 protein expression were measured by western blot, (F) the level of ROS expression was detected by ROS assay. **p < .01 and ***p < .001 vs. control (Ctrl) group. ##p < .01 and ###p < .001 vs. H2O2 group.
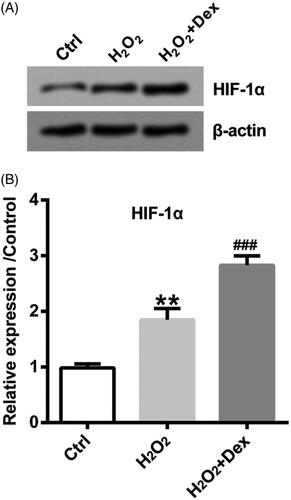
Dex induced the up-regulation of HIF-1α in PC12 cells
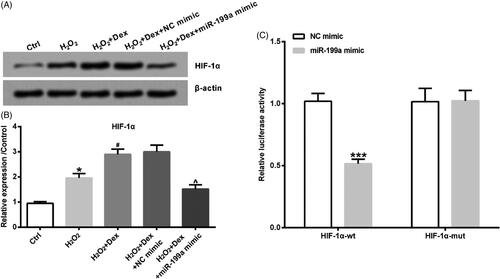
HIF-1α is a transcription factor regulated by oxygen. Next, the downstream effectors of which Dex exerted its function were explored. As depicted in , the level of HIF-1α was evidently up-regulated by H2O2, and H2O2-induced HIF-1α level was further raised by Dex in PC12 cells (both p < .05).
Figure 2. Dex induced the up-regulation of HIF-1α in PC12 cells. After pre-treatment by Dex (1 nM) for 30 min, cells were incubated by 50 µM H2O2 for 24 h. The level of HIF-1α protein was obtained by Western blot. **p < .01 vs. control (Ctrl) group. ###p < .001 vs. H2O2 group.
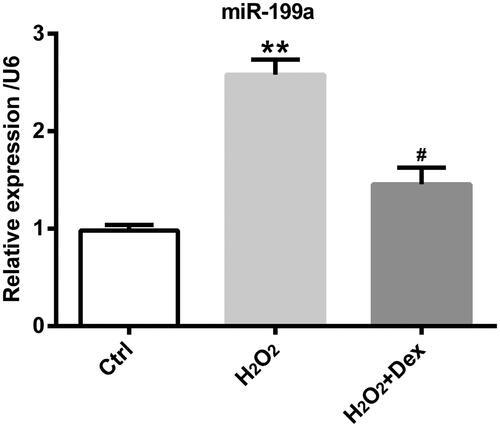
Dex decreased miR-199a levels in PC12 cells
As described in , we could get a massage that miR-199a expression was obviously increased by H2O2, but Dex could decrease miR-199a express evoked by H2O2 (both p < .05).
Figure 3. Dex decreased miR-199a levels in PC12 cells. After pre-treatment by Dex (1 nM) for 30 min, PC12 cells were incubated by H2O2 (50 µM) for 24 h. The levels of miR-199a expression were got by qRT-PCR. **p < .01 vs. control (Ctrl) group. #p < .05 vs. H2O2 group.
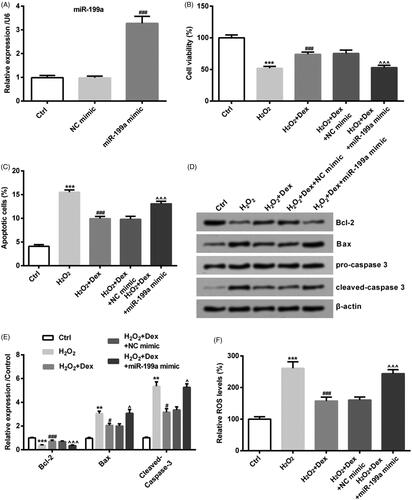
Dex attenuated H2O2-evoked oxidative damage by down-regulating miR-199a expression in PC12 cells
The NC mimic and miR-199a mimic were transfected into PC12 cells. After transfection, compared with the NC mimic transfected group, miR-199a level was significantly increased in the miR-199a mimic transfected group (p < .05, ). MiR-199a mimic could decrease cell viability induced by Dex (p < .05, ). MiR-199a mimic could reverse the decrease of cell apoptotic rate driven by Dex (p < .05, ). The changes of Bcl-2, Bax and cleaved-capase-3 protein level evoked by Dex were reversed by transfecting miR-199a mimic (all p < .05, ). The Dex-induced ROS level was increased by up-regulating miR-199a expression (p < .05, ).
Figure 4. Dex attenuated H2O2-evoked oxidative damage by down-regulating miR-199a expression in PC12 cells. NC mimic or miR-199a mimic was transfected in PC12 cells for 48 h. (A) The levels of miR-199a expression were computed by qRT-PCR. ###p< .001 vs. NC mimic group. After completing transfection by NC mimic or miR-199a mimic, PC12 cells were incubated by 50 µM H2O2 in combination with Dex (1 nM). (B) Cell viability was tested by CCK-8, (C) the rate of cell apoptosis was calculated by apoptosis assay, (D, E) the levels of Bcl-2, Bax and cleaved-caspase 3 proteins were measured by Western blot, (F) the level of ROS expression was detected by ROS assay. **p < .01 and ***p < .001 vs. control (Ctrl) group. #p < .05 and ###p < .001 vs. H2O2 group. ∧p < .05 and ∧∧∧p < .001 vs. H2O2+Dex + NC mimic group.
Dex up-regulated HIF-1α through declining miR-199a
As shown in , miR-199a mimic could down-regulate HIF-1α expression (p < .05). Moreover, as seen in , with overexpression of miR-199a, the assays disclosed markedly decline of luciferase reporter gene activity for the HIF-1α-wt-3′UTR constructs (p < .05). To the contrary, luciferase reporter gene activity was not changed when applying the HIF-1α-Mut-3′UTR.
Figure 5. Dex up-regulated HIF-1α through declining miR-199a. After transfection by NC mimic or miR-199a mimic, cells were incubated by 50 µM H2O2 in combination with Dex (1 nM). (A, B) The levels of HIF-1α protein were tested by western blot. *p < .05 vs. control (Ctrl) group. #p < .05 vs. H2O2 group. ∧p < .05 vs. H2O2 + Dex + NC mimic group. (C) Luciferase-containing HIF-1α 3′-UTR (HIF-1α-wt) or luciferase-containing Mut-HIF-1α 3′-UTR (HIF-1α-mut) and miR-199a mimic or NC mimic were transfected into PC12 cells. Relative luciferase activity was detected by dual luciferase activity assay. ***p < .001 vs. NC mimic group.
Dex activated PI3K/AKT/mTOR and Wnt/β-catenin pathways by down-regulating miR-199a
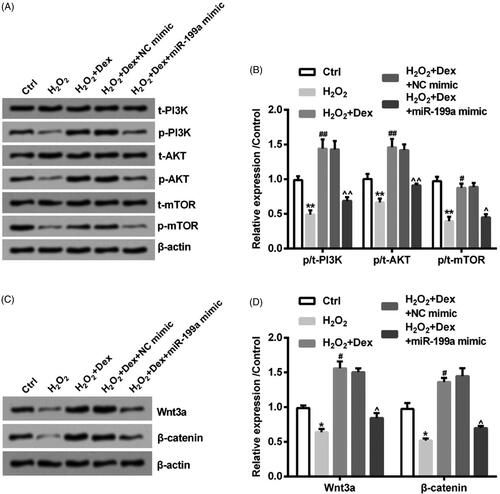
After miR-199a mimic and NC mimic were transfected, the simulation of PI3K/AKT/mTOR and Wnt/β-catenin pathways was tested by adopting western blot. The phosphorylation levels of PI3K, AKT and mTOR were decreased by H2O2 (all p < .05). The PI3K, AKT and mTOR phosphorylation levels were up-regulated by Dex (all p < .05). After transfection of miR-199a mimic, the increased phosphorylation levels of PI3K, AKT and mTOR induced by Dex were reversed (all p < .05, ). Furthermore, the Wnt3a and β-catenin expression was declined by H2O2 (both p < .05). The above two protein levels in H2O2-treated cells were increased by Dex (both p < .05). After transfection of miR-199a mimic, the two protein expression was decreased (both p < .05, ). Above all results demonstrated Dex induced the simulation of the above two pathways in PC12 cells through down-regulating miR-199a expression.
Figure 6. Dex activated PI3K/AKT/mTOR and Wnt/β-catenin pathways by down-regulating miR-199a. After transfection by NC mimic or miR-199a mimic, PC12 cells were incubated by 50 µM H2O2 in combination with Dex (1 nM). (A, B) The phosphorylation levels of PI3K, AKT, and mTOR were measured by Western blot. (C, D) The levels of β-catenin and Wnt3a expression were also tested by adopting Western blot. *p < .05 and **p < .01 vs. control (Ctrl) group. #p < .05 and ##p < .01 vs. H2O2 group. ∧p < .05 and ∧∧p < .01 vs. H2O2+Dex + NC mimic group.
Discussion
Oxidative stress could induce various diseases through oxidative modification of biomolecules and changes of signalling pathways including neurodegenerative diseases [Citation21]. Thus, the protection of cells from oxidative damage is critical for the treatment of neurodegenerative diseases. In our studies, H2O2 decreased viability and promoted apoptosis in PC12 cells. But, Dex raised cell viability and repressed apoptosis in PC12 cells. Furthermore, we found that Dex further up-regulated HIF-1α expression induced by H2O2. In addition, Dex up-regulated HIF-1α expression via reducing miR-199a in PC12 cells. It was tested that Dex attenuated H2O2-induced oxidative damage and induced the simulation of PI3K/AKT/mTOR and Wnt/β-catenin signalling pathways in PC12 cells through down-regulating miR-199a expression.
Currently, countless researches have showed that Dex exerts neuroprotective effects through different mechanisms [Citation3]. For instance, it was indicated that Dex could reverse neural apoptosis through anti-inflammatory and anti-apoptotic effects to protect neurons [Citation22]. There was also a study showed that Dex inhibited excitatory neurotransmitter and decreased calcium entry into cells, which ultimately attenuated cell damage [Citation23]. Besides, Dex reduced the release of glutamate and thus was considered to have neuroprotective effects [Citation24]. In addition, it was generally reported that Dex had neuroprotective effects in different animal models due to its anti-oxidative damage effects [Citation25–27]. This was similar to our results, in which Dex increased PC12 cell viability and decreased apoptosis, along with inhibited ROS formation to protect the cell from oxidative damage.
The HIF-1α controlled by oxygen was an ingredient that took part in the process of transcription and controlled the level of all kinds of genes [Citation28], which were involved in cell growth and apoptosis, like CyclinD1, p62, Bax and Bcl-2. In our studies, H2O2 increased HIF-1α level in PC12 cells and Dex further promoted the increase of HIF-1α level, thereby protected cells from oxidative damage. What is more, the levels of different miRNAs can drive the stabilization of HIF-1α. For example, miR-199, miR-20b together with miR-17-92 destabilized HIF-1α [Citation29,Citation30]. But miR-210 along with miR-424 stabilized HIF-1α [Citation31,Citation32]. Next, the downstream effectors of which Dex exerted its function were explored. H2O2 also induced the increase of miR-199a expression and Dex inhibited the expression of miR-199a. Hence, we speculated that Dex regulated HIF-1α might be through miR-199a. Dual-luciferase activity assay results showed that Dex up-regulated HIF-1α expression via the decline of miR-199a in PC12 cells and 3′-UTR of HIF-1α was as a specific target of miR-199a-5p.
MiRNAs were a glass of critical factors that participated in cell proliferation as well as apoptosis [Citation33]. A study has shown miR-199a-5p is strikingly declined in PD [Citation13]. Moreover, a study explained that miR-199a suppressed HIF-1α level under hypoxic environment [Citation34]. In addition to this, it was affirmed that the abnormal miR-199a expression was together with the generation of ROS [Citation35,Citation36]. Along with the deepened study gradually, a mass of researches released that miR-199a repressed cell viability and accelerated apoptosis [Citation37]. The above results of all researches were similar to our data. Although it was showed that miR-199a level took part in neurodegenerative disease, the specific neuroprotective mechanism of Dex was not clear. Hence, we collected PC12 cells to validate how the expression of miR-199a induced by Dex played a role in neurodegenerative diseases. Further results manifested that Dex-caused the decline of cell viability and the increase of apoptotic percentage through increasing the level of miR-199a. We additionally found that the increase of miR-199a resulted in ROS generation. The above all results illustrated that Dex could suppress oxidative damage driven by H2O2 via down-regulating miR-199a expression. Our study uncovered that Dex decreased the expression of miR-199a to play its neuroprotective role. There was a study released that the silence of long non-coding RNAs (lncRNA MALAT1) improved the protective effect of Dex on hypoxic-ischemic brain damage by sponging miR-429 [Citation38]. Another article also showed lncRNA SNHG12 could target miR-199a to ameliorate brain microvascular endothelial cell injury [Citation39]. Thus, we speculated that Dex possibly reduced miR-199a via the regulation of certain lncRNA (such as lncRNA SNHG12). Hence, we need further experiments to verify this.
The PI3K/AKT/mTOR and Wnt/β-catenin pathways took part in neurodegenerative diseases. For instance, simulation of the PI3K/AKT/mTOR pathway could inhibit cell apoptosis to mediate the neuroprotective effect in a cellular model of PD [Citation40]. Research also explained that the blockage of Wnt/β-catenin pathway was partially activated to protect nerve cells in PD and AD [Citation41,Citation42]. The report also explicated that miRNAs protected cell from damage via regulating the above two kinds of pathways in neurodegenerative diseases [Citation43,Citation44]. For instance, up-regulating miR-100 expression evoked the simulation of PI3K/AKT/mTOR pathway to decrease the incidence of PD [Citation43]. Down-regulating miR-214 expression induced the simulation of Wnt/β-catenin pathway to decline the rate of HD [Citation44]. Although a few studies tested the miRNAs expression regulated the above pathways in neurodegenerative diseases, related research is still insufficient. The study has exhibited that PI3K/AKT/mTOR signalling pathway is activated by miR-199a-5p inhibitor [Citation45]. This was similar to our experimental results. Our study released miR-199a overexpression could repress the mobilization of PI3K/AKT/mTOR and Wnt/β-catenin signalling pathways in PC12 cells. The focus of our study is on how Dex regulates those two pathways in PC12 cells. The data displayed H2O2 prevented the activation of the above two signalling pathways. Moreover, Dex induced the activation of those two pathways in PC12 cells via declining miR-199a expression.
In brief, this article depicted the effect of Dex on oxidative damage in PC12 cells. This study indicated that Dex could protect PC12 cells from oxidative damage. In addition, Dex attenuated H2O2-induced oxidative damage and induced the simulation of PI3K/AKT/mTOR and Wnt/β-catenin signalling pathways in PC12 cells through declining miR-199a expression. Although Dex has been used in clinical studies, the mechanism of Dex’s action is not fully known to this day. Our research put forward an idea that Dex exerted its neuroprotective function by reducing miR-199. This not only lays a better foundation for further study but also provides a theoretical basis for clinical application of Dex. In a clinical study, Dex acts as a neuroprotective drug from oxidative damage.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Radi E, Formichi P, Battisti C, et al. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis. 2014;42(s3):S125–S152.
- Salim S. Oxidative stress and the central nervous system. J Pharmacol Exp Ther. 2017;360(1):201–205.
- Sifringer M, von Haefen C, Krain M, et al. Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxid Med Cell Longev. 2015;2015:1–10.
- Xia R, Yin H, Xia ZY, et al. Effect of intravenous infusion of dexmedetomidine combined with inhalation of isoflurane on arterial oxygenation and intrapulmonary shunt during single-lung ventilation. Cell Biochem Biophys. 2013;67(3):1547–1550.
- Xiang H, Hu B, Li Z, et al. Dexmedetomidine controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Inflammation. 2014;37(5):1763–1770.
- Huang C, Ng OT, Chu JM, et al. Differential effects of propofol and dexmedetomidine on neuroinflammation induced by systemic endotoxin lipopolysaccharides in adult mice. Neurosci Lett. 2019;707:134309.
- Wang Y, Jia A, Ma W. Dexmedetomidine attenuates the toxicity of betaamyloid on neurons and astrocytes by increasing BDNF production under the regulation of HDAC2 and HDAC5. Mol Med Rep. 2019;19(1):533–540.
- Qiu L, Tan EK, Zeng L. microRNAs and neurodegenerative diseases. Adv Exp Med Biol. 2015;888:85–105.
- Juzwik CA, Drake SS, Zhang Y, et al. microRNA dysregulation in neurodegenerative diseases: a systematic review. Progress in Neurobiology. 2019;182:101664.
- Yoram Barak PG. Do social isolation and loneliness kill people with Alzheimer’s disease? OBM Geriatr. 2018;2(4).
- Liu W, Li L. MicroRNA expression profiling screen miR-3557/324-targeted CaMK/mTOR in the rat striatum of Parkinson’s disease in regular aerobic exercise. BioMed Res Int. 2019;2019:7654798.
- Lin Y, Liang X, Yao Y, et al. Osthole attenuates APP-induced Alzheimer’s disease through up-regulating miRNA-101a-3p. Life Sci. 2019;225:117–131.
- Tolosa E, Botta-Orfila T, Morato X, et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol Aging. 2018;69:283–291.
- Jiang Y, Zhu Y, Wang X, et al. Temporal regulation of HIF-1 and NF-kappaB in hypoxic hepatocarcinoma cells. Oncotarget. 2015;6(11):9409–9419.
- Joshi HP, Subramanian IV, Schnettler EK, et al. Dynamin 2 along with microRNA-199a reciprocally regulate hypoxia-inducible factors and ovarian cancer metastasis. Proc Natl Acad Sci USA. 2014;111(14):5331–5336.
- Mizuno S, Bogaard HJ, Gomez-Arroyo J, et al. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1alpha expression in lungs from patients with COPD. Chest. 2012;142(3):663–672.
- Raimondi L, Amodio N, Di Martino MT, et al. Targeting of multiple myeloma-related angiogenesis by miR-199a-5p mimics: in vitro and in vivo anti-tumor activity. Oncotarget. 2014;5(10):3039–3054.
- Pislar AH, Zidar N, Kikelj D, et al. Cathepsin X promotes 6-hydroxydopamine-induced apoptosis of PC12 and SH-SY5Y cells. Neuropharmacology. 2014;82:121–131.
- Pasban-Aliabadi H, Esmaeili-Mahani S, Sheibani V, et al. Inhibition of 6-hydroxydopamine-induced PC12 cell apoptosis by olive (Olea europaea L.) leaf extract is performed by its main component oleuropein. Rejuvenat Res. 2013;16(2):134–142.
- Hu X, Zhang H, Zhang Y, et al. Differential protein profile of PC12 cells exposed to proteasomal inhibitor lactacystin. Neurosci Lett. 2014;575:25–30.
- Thanan R, Oikawa S, Hiraku Y, et al. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int J Mol Sci. 2014;16(1):193–217.
- Ning Q, Liu Z, Wang X, et al. Neurodegenerative changes and neuroapoptosis induced by systemic lipopolysaccharide administration are reversed by dexmedetomidine treatment in mice. Neurol Res. 2017;39(4):357–366.
- Engelhard K, Werner C, Kaspar S, et al. Effect of the alpha2-agonist dexmedetomidine on cerebral neurotransmitter concentrations during cerebral ischemia in rats. Anesthesiology. 2002;96(2):450–457.
- Chen Y, Zhao Z, Code WE, et al. A correlation between dexmedetomidine-induced biphasic increases in free cytosolic calcium concentration and energy metabolism in astrocytes. Anesth Analg. 2000;91(2):353–357.
- Cosar M, Eser O, Fidan H, et al. The neuroprotective effect of dexmedetomidine in the hippocampus of rabbits after subarachnoid hemorrhage. Surg Neurol. 2009;71(1):54–59; discussion 59.
- Duan X, Li Y, Zhou C, et al. Dexmedetomidine provides neuroprotection: impact on ketamine-induced neuroapoptosis in the developing rat brain. Acta Anaesthesiol Scand. 2014;58(9):1121–1126.
- Xiong B, Shi QQ, Miao CH. Dexmedetomidine renders a brain protection on hippocampal formation through inhibition of nNOS-NO signalling in endotoxin-induced shock rats. Brain Inj. 2014;28(7):1003–1008.
- Im S, Kim DW. Nkx3.2 induces oxygen concentration-independent and lysosome-dependent degradation of HIF-1alpha to modulate hypoxic responses in chondrocytes. Cell Signal. 2017;36:127–138.
- Rane S, He M, Sayed D, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104(7):879–886.
- Lei Z, Li B, Yang Z, et al. Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLOS One. 2009;4(10):e7629.
- Kelly TJ, Souza AL, Clish CB, et al. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol Cell Biol. 2011;31(13):2696–2706.
- Ghosh G, Subramanian IV, Adhikari N, et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest. 2010;120(11):4141–4154.
- Ghaemi Z, Soltani BM, Mowla SJ. MicroRNA-326 functions as a tumor suppressor in breast cancer by targeting ErbB/PI3K signaling pathway. Front Oncol. 2019;9:653.
- Zhang H, Zhu S, Zhang C, et al. miR-199a-5p inhibits the proliferation of rat airway smooth muscle cells and the expression of hypoxia inducible factor 1 alpha under hypoxia conditions. Xi Bao yu Fen zi Mian yi Xue za Zhi [Chin J Cell Mol Immunol]. 2015;31(9):1183–1188.
- Lin J, Chuang CC, Zuo L. Potential roles of microRNAs and ROS in colorectal cancer: diagnostic biomarkers and therapeutic targets. Oncotarget. 2017;8(10):17328–17346.
- He J, Wang M, Jiang Y, et al. Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1alpha/COX-2 pathway. Environ Health Perspect. 2014;122(3):255–261.
- Wang H, Wang Z, Tang Q. Reduced expression of microRNA-199a-3p is associated with vascular endothelial cell injury induced by type 2 diabetes mellitus. Exp Ther Med. 2018;16(4):3639–3645.
- Fang H, Li HF, He MH, et al. Long non-coding RNA MALAT1 sponges microRNA-429 to regulate apoptosis of hippocampal neurons in hypoxic-ischemic brain damage by regulating WNT1. Brain Res Bull. 2019;152:1–10.
- Long FQ, Su QJ, Zhou JX, et al. LncRNA SNHG12 ameliorates brain microvascular endothelial cell injury by targeting miR-199a. Neural Regen Res. 2018;13(11):1919–1926.
- Nakaso K, Ito S, Nakashima K. Caffeine activates the PI3K/Akt pathway and prevents apoptotic cell death in a Parkinson’s disease model of SH-SY5Y cells. Neurosci Lett. 2008;432(2):146–150.
- Cao W, Dong Y, Zhao W, et al. Mulberrin attenuates 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-induced Parkinson’s disease by promoting Wnt/beta-catenin signaling pathway. J Chem Neuroanat. 2019;98:63–70.
- Hu Y, Chen W, Wu L, et al. TGF-beta1 restores hippocampal synaptic plasticity and memory in Alzheimer model via the PI3K/Akt/Wnt/beta-catenin signaling pathway. J Mol Neurosci. 2019;67(1):142–149.
- Peng T, Liu X, Wang J, et al. Long noncoding RNA HAGLROS regulates apoptosis and autophagy in Parkinson’s disease via regulating miR-100/ATG10 axis and PI3K/Akt/mTOR pathway activation. Artif Cells Nanomedicine Biotechnol. 2019;47(1):2764–2774.
- Ghatak S, Raha S. Micro RNA-214 contributes to proteasome independent downregulation of beta catenin in Huntington’s disease knock-in striatal cell model STHdhQ111/Q111. Biochem Biophys Res Commun. 2015;459(3):509–514.
- Chen J, Gong X, Huang L, et al. MiR-199a-5p regulates sirtuin1 and PI3K in the rat hippocampus with intrauterine growth restriction. Sci Rep. 2018;8(1):13813.