
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Kojic acid (KA) as tyrosinase inhibitor shows insufficient skin penetration and several adverse events due topical administration. KA solid lipid nanoparticles (KA-SLNs) were prepared using high speed homogenisation followed by ultra-probe sonication method for improve its effectiveness.
KA-SLNs was optimised by Glyceryl mono-stearate (GMS) and Cholesterol (Chol) as lipid excipients and span 60 (SP 60) and Tween 20 (Tw 20) as co-emulsifiers (particle size 156.97 ± 7.15 nm, encapsulation efficiency 59.02 ± 0.74%, drug loading 14.755 ± 1.63%, polydispersity index (PDI) of 0.388 ± 0.004 and zeta potential (ZP) of -27.67 ± 1.89 mV). Optimum formulation (KA-SLN3 dispersion) was stable at 4 and 25 °C for 3 months. Also, TEM image confirmed these results. The results of XRD, DSC and ATR-FTIR analysis indicated that KA was well encapsulated within the SLNs either in molecularly dispersed state and stabilised in amorphous form and there was no chemical interaction between drug and other ingredients. Controlled release was achieved with this formulation. KA-SLN3 dispersion have more tyrosinase inhibition potency in comparison with pure KA. Also, the results of the ex vivo and in vitro percutaneous absorption show that KA-SLN3 dispersion improved percutaneous delivery of KA as a promising and potential novel topical preparation and might open new avenues for treatment of hyperpigmentation disorders.
Introduction
Cutaneous melanin (an irregular light-absorbing polymer) is a ubiquitous class of biological pigments [Citation1]. The step of melanin synthesis in melanocytes is carried out through the complex chemical process by several enzymes. Tyrosinase (monophenol or o-diphenol oxygen oxidoreductase) is an essential enzyme for melanin synthesis that is a copper-containing enzyme. In addition, it has been introduced as polyphenol oxidase, phenolase, catecholase and cresolase [Citation2,Citation3].
Pigmentation disorders are one of the most common skin diseases that are caused by abnormal distribution, structure and function of melanocytes. The pigmentation disorders’ treatment has always been challenging and discouraging for the most population especially women. The most middle-aged women have often abnormal pigmentation on their facial skin and these skin disorders are related to various endogenous and exogenous agents [Citation4]. Pigmentation disorders can be expressed in two forms of skin pigmentation increase (hyperpigmentation) and decrease (hypopigmentation). These skin pigmentation disorders can cause aesthetic, cognitive and psychological challenges for the patients and have psychosocial effects on them. Common typical pigmentation disorders including melasma (chloasma) and ephelides (freckles) are related to disturbance in skin pigmentation [Citation5–7].
Some of skin lighteners can inhibit the synthesis of melanin; therefore, they can be considered as a therapeutic agent for hyperpigmentation disorders. KA and its derivatives act as tyrosinase inhibitors in topical products which is derived from mushrooms natural metabolite. It is obtained from various types of fungi such as Aspergillus, Penicillium, Alternaria, Pleospora, Syncephalastrum, Fusarium, Mucor, etc. KA as a hydrophilic ingredient led to inadequate tyrosinase inhibitor activity in vivo. KA concentration is usually 1–4% in skin lightening products [Citation2,Citation8–10]. Due to insufficient skin penetration ability, skin irritation, cytotoxicity and instability on storage, KA applications in dermal preparations has been limited [Citation2,Citation11–13].
Development of delivery systems for hydrophilic drugs is very important for the following reasons: (a) the importance of investigations in the field of macromolecules such as proteins, peptides and nucleic acids in the treatment of various diseases, (b) the increasing interest in treatment with hydrophilic drugs [Citation14,Citation15]. But there are major challenges that we are faced in delivering macromolecules and hydrophilic drugs as follows: (a) unsuitable biodistribution and low permeability across barriers, (b) low bioavailability and hydrolytic degradation, (c) a short biological half-life in the circulatory system and unsuitable pharmacokinetics. To overcome this problem, nanomedicines have been developed for encapsulate and targeting hydrophilic drugs [Citation16].
Recently, solid lipid nanoparticles (SLN) have been proposed as new drug delivery systems for topical and percutaneous preparations. Encapsulating hydrophilic drugs in SLNs have shown promising results as potential drug vehicles for targeted co-delivery or stimuli-responsiveness due to the better characteristics such as small particle size with relatively narrow size distribution, facilitate drug permeation through breaking the biological barriers, possible sterilisation by gamma irradiation and autoclaving, avoidance of organic solvents, enhanced pharmacokinetic properties and resistance to chemical degradation, provide controlled release of drugs through the skin epidermis, possible production no toxic metabolites improved drug bioavailability and the possibility of surface modification, reduce the adverse events, possible spray dried and lyophilisation, improve skin hydration, control systemic absorption and the stabilise sensitive compounds against light and hydrolysis and ease of scale-up and manufacturing [Citation17–19].
During the past years, some of hydrophilic tyrosinase inhibitor agents have been used in the treatment of pigmentation disorders which includes the following: KA and its derivatives, glycolic acid (GA), hydroquinone (HQ), and vitamin C (VC), arbutin, etc. [Citation10]. Among them, several claims such as coated liposomes loaded with KA, KA-polymer-based magnetic nanocomposites, hydroquinone-SLNs, liposomes loaded with arbutin and vitamin C polymeric nanoparticles have been investigated as novel drug delivery systems for improving their effectiveness [Citation20–24].
The aim of the current study is to evaluate the role of various formulation factors i.e. the type of lipid and mixture of surfactants on physicochemical properties of KA-SLNs which could potentially be influenced particle size, polydispersity index (PDI), zeta potential, entrapment efficiency (EE), morphology, crystal structures, release behaviour of KA nanocarriers and percutaneous absorption.
Materials and methods
Materials
KA and Palmitic acid (PA) were purchased from Sigma-Aldrich. Tween 20, Span 80, stearic acid (SA) and cholesterol (Chol) were obtained from Merck (Merck Co., Germany). Span 60 was received from Daejung (Daejung Chemicals & Metals Co., Ltd. Korea). Beeswax (BW) was obtained from John’s Laboratory (John’s Laboratory Chemicals, India). Glyceryl monostearate (GMS) was gifted by Gattefossé (France). Deionised water was purified using a Human power 2 system (human Co., Korea). Mushroom tyrosinase (EC1.14.18.1), L-3,4-dihydroxyphenylalanine (L-DOPA) were from Sigma Chemical Co. (St. Louis, MO, USA).
Preparation of KA-SLNs
KA-SLNs were prepared by probe sonication (ultrasonication) technique [Citation25]. The water phase and lipid phase were prepared according to the following procedure: A mixture of GMS, Chol and Span® 60 was heated at 95 °C (lipid phase); Deionised water (DIW) and Tween® 20 were homogenised using a high-shear homogeniser (D-91126 Schwabach, Heidolph, Germany) and then was heated at 95 °C (the water phase). One-third of the water phase (95 °C) containing Tween® 20 placed in an ice-water bath. A mixture of KA and two-third of the preheated water phase (95 °C) was added dropwise to the lipid phase and it was immediately sonicated using a high intensity probe sonicate (Bandelin, 3100, Germany) for 5 min (100% amplitude). After sonication, the remained one-third of the water phase in an ice-water bath was added into the mixture to form an initial pre-emulsion and all of the mixture quickly was placed in an ice-water bath. The mixture was again subjected to sonication for 5 min transferred to ice bath finally. to high intensity probe sonication (100% amplitude) for 30 min.
Determination of physicochemical characterisation
Particle size and Zeta potential
The mean particle size (as the hydrodynamic diameter), size distribution (polydispersity index, PDI) and zeta potential (electrophoretic mobility) of KA-SLNs were characterised by dynamic light scattering (DLS), using a Zetasizer Nano ZS (Malvern Instruments, UK) with a laser Doppler velocimetry at 25 °C and a 90° detector angle [Citation26]. Each sample was performed in 14 runs and was repeated three times and the results finally were reported as mean ± SD ().
Table 1. Component and physicochemical properties of investigated KA-SLNs (% w/w). The data are the mean and standard deviation of three determinations (n = 3).
Entrapment efficiency (EE)
EE percentage values of KA-SLNs is the percentage of trapped KA ratio to the initially drug. It was considered by determining the amount of free KA (non-encapsulated in KA-SLNs) in the aqueous surfactant solution. EE% of KA-SLNs were determined by first separation of the non-encapsulated KA using a centrifuge method (Sigma 3-30KS, Germany) at 462893.4 RCF (RCF Relative Centrifugal Force) for 20 min at 4 °C. The amount of KA in supernatant was determined using a UV-visible spectrophotometer (JASCO V-630) method (λ max of KA: 268.5 nm). EE and DL (drug loading) percentage values of KA-SLNs were calculated by the following equation, EquationEquations (2)(2)
(2) and Equation(3)
(3)
(3) :
(1)
(1)
(2)
(2)
(3)
(3)
where WInitial drug is total amount of KA used in the formulation and WFree drug is the amount of drug detected in supernatant after centrifugation of the KA-SLNs formulation [Citation27–29].
Attenuated total reflectance-Fourier transform infra-red spectroscopy (ATR-FTIR)
Optimum formulation of KA-SLNs were lyophilised in −80 °C for 30 h and then lyophilised in −50 °C and 0.1 mbar (Marin Christ, Osterode, Germany). The KA, freeze-dried KA-SLN3, native lipid, and physical mixture of optimum formulation components were analysed to identify their intermolecular interactions by infra-red spectra, using attenuated total resonance (ATR) FTIR spectrophotometer (Agilent CARY 630 ATR-FTIR) [Citation30]. A specimen of each component was included on the diamond ATR crystal and Agilent resolutions pro-software was utilised to analyse and scan the spectra. Each ATR-FTIR spectrum represented an average of 32 scans at a resolution of 4 cm−1. The ATR-FTIR spectra were recorded in the range between 3800 cm−1 and 700 cm−1.
Transmission electron microscope (TEM) analysis
TEM (912AB, Leo, England) imaging method was performed for characterising the structure and morphology properties, study crystal structures, symmetry, orientation and crystalline defects of the KA-SLNs formulations [Citation31]. Samples of KA-SLN3 were placed on copper grids and dried. Samples surface of KA-SLN3 was dried after being stained with a drop of 1% aqueous solution of uranyl acetate for contrast enhancement. Samples KA-SLN3 assessed by TEM after the samples were completely dried at room temperature.
Differential scanning calorimetry (DSC) analysis
DSC technique was examined to analysis the matrix structure and thermal behaviours studies of KA-SLNs [Citation32]. Therefore, to ensure the encapsulation of KA in SLNs, the DSC method was employed. The freeze-dried samples used for ATR-FTIR studies were employed for DSC analysis. Five milligrams of KA, the freeze-dried KA-SLN3, native lipid, physicochemical mixtures of optimum formulation components, were placed in an aluminium oxide pan, respectively. Then the specimens were sealed inside the pans and an empty pan was applied as control. The samples with DSC (Pyris 6, PerkinElmer, USA) method were scanned at 20–300 °C temperature range by calorific rate of 20 °C/min under N2 flow. Eventually, the obtained results of the DSC thermograms were used for evaluation drug-excipient interactions.
Powder X-ray diffraction (PXRD)
PXRD method was performed on the samples for characterising creation changes in the crystal lattice structure of component parts after the preparation of the KA-SLNs formulations [Citation33]. Also, XRD data confirmed the results demonstrated by DSC study. The samples were used for PXRD by Bruker D8 Advance X-ray diffractometer (Germany) (40 kV and 30 mA) analysis too. The encapsulation of KA inside the SLNs was further confirmed by the obtained diffractograms of PXRD. Samples were exposed to Cu Kα radiation (λ = 0.15406 nm) that it equipped with a sample spinner operating at a current of 30 mA and a voltage of 40 kV. The measurements were done at room temperature and running at 2θ from 1° to 100°, with a 0.04° step size and 1 s step time.
Drug release
The release profile of drugs in SLNs is a main factor in determining its efficacy [Citation34,Citation35]. The drug loaded nanoparticles were dispersed in dissolution media (Phosphate buffer, pH 6.8) and then samples of the media were removed at certain profile intervals to measure concentration of the percent of drug released using UV-spectroscopy. The release profile of KA-SLN3 was determined by paddle method (type II) mentioned in United States Pharmacopoeia (USP XXVIII, 2005). The Erweka dissolution apparatus (DT620, Erweka, Germany) was utilised to carry out the drug release tests.
About 900 ml of dissolution medium was prepared for the release test at 37.0 °C and 60 rpm was defined as the rotation speed. Then, the samples (5 ml) were removed from the dissolution media at various time intervals (0.5, 1, 2, 4, 6, 8 and 24 h) and replaced with fresh medium at the same temperature and the amount of KA in samples were assayed using the UV method at 268.5 nm.
Determination of mushroom tyrosinase activity
Tyrosinase from mushroom (EC number 1.14.18.1, Sigma T3824-25KU, 2680 units/mg solid) applied for the bioassay of KA-SLN3 and KA solution according to previous studies with slight modifications by M. Khoshneviszadeh et al. [Citation36,Citation37]. Mushroom tyrosinase inhibitory activity was evaluated using L-DOPA as a substrate and the formation of dopachrome was observed at 475 nm [Citation38,Citation39]. All test samples were first dissolved in phosphate buffer (pH 6.8) at 40 mM and diluted to the required concentrations (KA-SLN3: 10,00,50,02,50,25,02,00,00,00,000 µg/ml and KA: 1,00,50,25,105 µg/ml). First, 10 µL of tyrosinase (0.5 mg/mL) were mixed with 160 µL of 50 mM phosphate buffer (pH 6.8) and then 10 µL of the test sample were added to 96-well microplates. After pre-incubation at 28 °C for 20 min, 20 µL of L-DOPA solution (0.5 mM) were added to the phosphate buffer and KA was used as the positive control. Each assay was conducted as three separate replicates. The inhibitory effects of the tested compounds were expressed as the concentrations that inhibited 50% of the enzyme activity (IC50). The percent inhibition ratio was calculated according to the following equation:
(4)
(4)
Ex vivo and in vitro skin permeation/penetration study
Percutaneous absorption was evaluated via rat skin, human skin samples ex vivo and synthesis membrane as artificial skin [Citation40]. Intact human skin samples were donated from the abdominal region of a young female in Sari Pars Clinic Surgery Centre. The segments of human skin, included epidermis, were stored in PBS solution before study and subcutaneous tissue samples were taken by blunt scissors and were cut into 2 × 2 cm pieces with a 2 mm thickness. Alcohol was used to clean and remove possible contaminations of subcutaneous lipid.
The rat skin was obtained from 1.5-month male Wistar rats with weighing 200–250 g. The hairs of rat skin were carefully and mildly shaved using electric hand razors before experiments and then the rats were killed by chloroform inhalation after 48 h. Full-thickness of the rat skin was surgically removed from the abdominal region and animal skin samples were cut into 2 × 2 cm slices with a 1 mm thickness. Separated rat skins were placed within a normal saline solution for 24 h before starting the diffusion assay to remove adhering lipid subcutaneous layer, extraneous debris and leachable enzymes of the skin samples. All experimental and surgical approaches were assessed and approved by the animal and ethics review committee for Animal Experimentation of Mazandaran University of Medical Sciences (IR.MAZUMS.REC.96-10322) Sari, Iran.
Strat-M® Membrane, as artificial skin, was purchased from Merck (Merck Co., Germany). Strat-M® Membrane is a non-animal and synthetic skin based in vitro model for percutaneous absorption tests that is designed for predictive of drug diffusion in human skin and is a safety testing for cosmetic formulations and personal care products and active pharmaceutical ingredients and it is without storage limitations and lot-to-lot variability.
Rat skin, human abdominal skin and synthesis membrane (as artificial skin) were applied for study the percutaneous penetration of 3 ml of selected formulation of KA-SLN3 dispersion in comparison with KA solution as donor phase (n = 3). Receptors phase (at the dermis side) was filled by 33 ml of phosphate buffer (PBS, pH 7.4) and cells were set at 37 ± 0.5 °C with the stirring rate at 600 rpm [Citation41].
The skin surface temperature was set to 32 °C for in vivo conditions simulation. A 5 ml aliquot was withdrawn from the receptor medium at pre-determined time intervals (1, 2, 3, 4, 6, 8, 12 and 24 h) and then replaced with an equal volume of medium.
The skins were removed from Franz cell at the end of study (after 24 h). Skin samples washed three times with PBS, pH 7.4 for removing the remaining materials and cut into small pieces, were placed in a buffer solution and the samples were sonicated for 1 h using a bath sonicator. Supernatants were collected and filtered by 0.22 µm membrane and quantified using a spectrophotometer device at a 268 nm for KA content.
Stability studies
Stability studies [Citation42] of the developed KA-SLN3 were performed at two different temperature conditions at 4 °C and at room temperature (RT) for a period of 3 months. The physical stability of KA-SLN3 were monitored to determine the size, PDI and ZP and physical appearance such as change in the colour of the formulation and formation of aggregation of lipid excipients and or precipitate.
Statistical analysis
The results of experimental data are illustrated as mean ± SD of at least three determinations (n = 3). Statistical analysis performed using one sample t-test and one-way analysis of variance (ANOVA) followed by Tuckey–Kramer multiple comparison post-tests (using the SPSS 2021 software) and p value < .05 was considered as the minimal level of significancy.
Results and discussion
KA-SLN characteristics (size, PDI, zeta potential and entrapment effciency) analysis
Selection of suitable lipid/s for KA-SLNs preparation was the main stage for achieving proper characteristics (particle size, PDI, zeta potential and drug loading) in KA nanoparticles. Several lipids and surfactant mixtures were investigated for KA-SLN formulation due to highly water solubility of KA (). KA-SLNs were prepared using high speed homogenisation followed by ultra-probe sonication method. Pre-formulation studies of the lipid excipients were done on the basis of suitable characteristics of the KA-SLNs in several formulations containing various lipids such GMS, SA, Chol, BW, PA and tween 20, span 60 and span 80 were selected as surfactants. The contents of KA, surfactants and lipids were kept constant, as the goal was to obtain maximum EE% in SLNs.
In our previous studies, Rostamkalaei et al. demonstrated that the presence of cholesterol in the lipid matrix of SLNs decreased the crystallinity of the lipid phase and increased drug encapsulation in SLNs. In pre-formulation studies, the particle size of KA-SLNs decreased by the addition of Span 60 as co-emulsifier according to Rostamkalaei et al. [Citation29]. Results of pre-formulation studies revealed the highest entrapment in the SLNs containing span 60 (it has the highest transition temperature) in comparison with span 80 according to Kelidari et al. [Citation43]. Kiran Yadav et al. showed that the presence of very high cholesterol content in the lipid matrix of lipid nanoparticles had a lowering effect on drug entrapment [Citation44].
Initial attempts on pre-formulation studies revealed that speed and time of stirrer, temperature, frequency of probe sonicator homogenisation cycles, time sonicate had affected on the mean particle size, PDI, EE% and zeta potential of the SLNs. Higher stirring speeds prevent the particle aggregation because of kinetic energy and high shear. The stirrer speed from 3000 to 8000 rpm for 10 min produced the smallest particle size because more increase in the stirring rate and time had minor effect on the KA-SLN characteristics. Number of homogenisation cycles and frequency of probe sonicator were evaluated by doing the experiments at 30, 50, 75, 90 and 100% amplitude for two cycles and finally maximum reduction of mean particle size was achieved at 100% amplitude.
Lipid matrix of GMS and Chol cause a decrease in particle size, PDI and an increase in zeta potential and higher value of %EE compared to lipid matrix of Chol and others lipids such as SA, BW, PA separately. GMS and Chol were selected as the best excipients to prepare KA-SLN. The results revealed suitable physicochemical properties in the lipid matrix containing GMS and Chol in comparison with other investigated lipids.
Table one shows the characteristics of investigated KA-SLNs. These results indicate the lowest particle size in formulations containing GMS and Chol in comparison with other lipids (p < .001). The composition of surfactants had no effect on mean particle size in KA-SLN3, 4 formulations containing GMS (p > .05). These nanoparticles characterised by the highest zeta potential too (p < .001). These data show low mean particle size and high ZP in KA-SLN3 and KA-SLN4.
The results of drug loading and dispersity of particle size showed no significant differences between investigated formulations (p > .05). It seems that the kind of lipid matrix and surfactant mixture had no effect on EE and PDI in these formulations.
Several studies have shown that the particle size of drug loaded SLNs has been affected by type of lipid, chemical and physical structure of solid lipid carrier, polymorphic state of lipid excipients, the composition of surfactants and the method of preparation, etc. [Citation45]. EE values of these nanoparticles might be affected by drug miscibility and solubility within melted lipid, physical and chemical structure of solid lipid, polymorphic state of lipid ingredients [Citation46].
Particle size, size distribution or polydispersity index (PDI) and zeta potential are used for determining the characterisations of KA-SLNs. These parameters mainly depend upon the preparation method and the ingredient composition. Hydrodynamic diameter of the SLNs (the z-average diameter or intensity weighted mean diameter) characterises by DLS and the PDI presents as an index of the width of the particle size distribution.
The PDI value that shows the quality of the dispersion ranges from 0 to 1. PDI values ≤0.1 represent the highest quality dispersion of nanoparticles. Most investigators characterised that dispersion ranges of PDI ≤0.3 are optimum values and PDI values ≤0.5 are suitable [Citation47].
The sizes of KA-SLNs formulations were between 156.97 to 392.13 nm. The PDI ranges in these investigated nanoparticles were between 0.259 − 0.424 and show suitable criteria; Further PDI results was characterised as reported by Bazzaz et al. [Citation48].
Zeta potential is index of the degree of repulsion between similarly charged particles in the nano systems. Polarity of particles can be either positive or negative that it represents the electric charge on the particle surface. physical stability of nanoparticles is expressive repulsive forces between nanoparticles which prevents the accumulation of nanoparticles during storage [Citation49].
The zeta potential of KA-SLNs showed several amounts between −3.18 and −27.67 mv. Bhalekar et al. showed that different formulations of lipids influencing the zeta potential of SLNs [Citation50]. EE values of drug-SLNs affected by drug miscibility and solubility within melted lipid, physical and chemical structure of solid lipid, polymorphic state of lipid ingredients [Citation46]. KA-SLNs showed drug loading between 45.6% and 59.02%. These entrapments are suitable for a hydrophilic drug loading in lipid nanoparticles. These results of EE% for a hydrophilic drug with typical property agree with the findings of Liu et al. [Citation51].
In other studies of hydrophilic drugs, Jain et al. showed that different combinations of GMS influencing the size of SLNs. In fact, they indicated that 1:1 ratio of GMS/Gelucire cause a decrease in particle size, PDI and an increase in zeta potential and higher value of %EE. Also, they have been reported that high entrapment efficiency of GMS/Gelucire is related to more solubility of drug in the GMS/Gelucire [Citation52]. Also, V. S. Shenoy et al. showed that the paclitaxel (PCL) SLNs prepared from GMS have a decrease in particle size, PDI and an increase in zeta potential and high entrapment efficiency using modified hot homogenisation [Citation53].
In another study, Kumar et al. indicated that GMS is suitable for the preparation of mefenamic acid (MF) SLNs compared to palmitic acid and stearic acid. Because MF has the most solubility in GMS than other above mentioned lipids. Particle size, PDI, zeta potential and EE% of drug were determined 109.7 nm, 0.34, 20.3 mV and 75.45 respectively [Citation54].
Transmission electron microscopy (TEM)
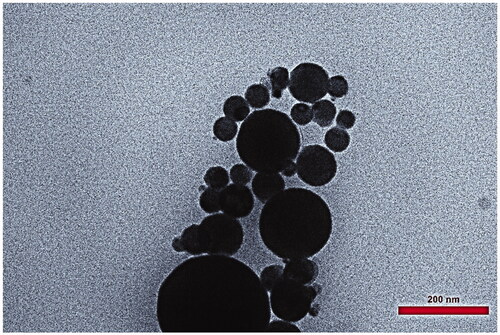
(KA-SLN3) represented TEM image of the morphology, particle shape, size and Zeta potential were measured by laser of DLS analyser. showed that the nanoparticles had nano size spherical forms and a narrow size distribution. TEM image showed that mixture of SP 60 and lipid matrix (GMS and Chol) had produced stable KA-SLN3.
Figure 1. TEM micrographs of KA-SLN (KA-SLN3).
Attenuated total reflectance-Fourier transform infra-red spectroscopy (ATR-FTIR)
ATR-FTIR studies were performed to find out any chemical interaction between KA and the different components of SLN. The ATR-FTIR spectra of KA solution, GMS, Chol, KA-SLN3 and physical mixture with the same composition have been shown in .
Figure 2. The ATR-FTIR spectra of KA, Chol, GMS, physical mixture and KA-SLN3.
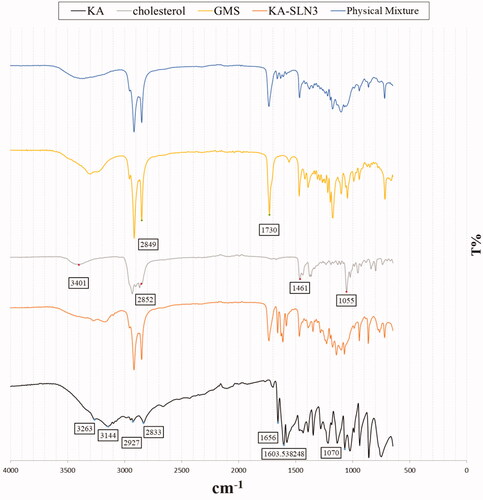
The ATR-FTIR spectrum of KA represented absorption bands at 3263 cm−1 and 3144 cm−1 due to O-H groups stretching vibrations, also peaks at 2925 cm−1 and 2833 cm−1 (aliphatic C-H stretching), 1656 cm−1 (C=O stretching of ring), 1607 cm−1 (C=C stretching), and 1069 cm−1 (C–O stretching of ring).
The ATR-FTIR spectrum of Chol showed an absorption band at 3402 cm−1 (O–H stretching), the bands at 3000–2850 cm−1 due to C–H of CH2 and CH3 groups, asymmetric and symmetric stretching vibrations, and a peak at 1054 cm−1 (C–O stretching vibration). In addition, the bands were observed at 1463–1457 cm−1, which were related to C–H bending vibrations.
The ATR-FTIR spectrum of GMS revealed absorption bands at 3400–3200 cm−1 corresponding to O–H stretching and the bands at 3000–2850 cm−1 due to C–H stretching vibrations, also a peak was observed at 1730 cm−1 (C=O stretching). Moreover, ATR-FTIR studies on GMS showed peaks at 1300–1000 cm−1, which were related to C–O stretching vibrations.
On the basis of ATR-FTIR analysis of physical mixture and KA-SLN3 formulation, it demonstrated that there was no shift in the characteristic peaks related to C=O and C=C groups of KA. Thus, it confirmed that there was no chemical interaction between drug and other formulation components.
Differential scanning calorimetry (DSC) and XRD
DSC thermograms was applied to determine the degree and state of crystallinity lattice of lipid dispersions, semi-solid systems and finding of the melting point and crystallisation form of substances such as SLNs [Citation55]. The KA, GMS, Chol, the physical mixture and KA-SLN3 powder were assayed by DSC (). KA and Chol displayed a single sharp endothermic melting peak at 158.58 °C and 149.10 °C respectively according to their melting points, a sign of intensely crystalline character for these two substances. Also in another study by S.V.K. Rompicharla et al., the Chol melting temperature was reported at 150.25 °C indicating its crystalline nature [Citation56].
Figure 3. DSC thermograms of KA, Chol, GMS, physical mixture and KA-SLN3.
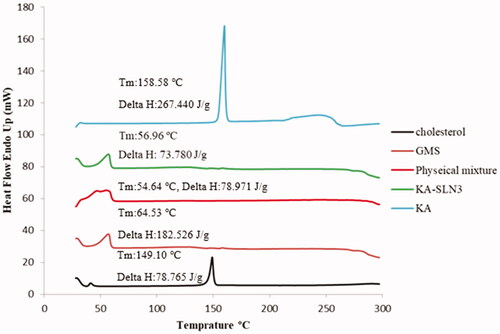
In this study, the thermograms of solid lipid (GMS) showed a single sharp endothermic melting peak around 60 °C according to these results [Citation57,Citation58]. The DSC thermograms of KA-SLN3 and physical mixture showed the endothermic peak around GMS melting point, but the endothermic peak of KA was not seen in KA-SLN3 lyophilised and physical mixture. This thermograms indicate that the physical state of KA changed from crystalline to amorphous form (). Other reports show drug loaded SLN was present in amorphous form and may have been homogeneously dispersed in the lipid matrix too [Citation29].
XRD diffractograms of the lipid (GMS and Chol), pure KA, the freeze-dried KA-SLN3 and physical mixture of optimum formulation components are shown in . XRD spectra of the Chol appeared the peaks at 2θ: 5.1235°, 6.226°, 6.89°, 7.61°, 9.410°, 10.415°, 11.15°, 11.554°, 12.69°, 13.906°, 15.272°, 16.822°, 17.30°, 17.958°, 18.71°, 19.22°, 20.93°, 23.374°, 25.20° according to Rostamkalaei et al. [Citation29]. Also, characteristic sharp peaks (XRD pattern of GMS) were observed in at 2θ: 5.412°, 19.299°, 20.27°, 21.35°, 22.68°, 23.208°, 36.57° according to Kumar et al. [Citation59].
Figure 4. XRD of KA, Chol, GMS, physical mixtures and KA-SLN3.
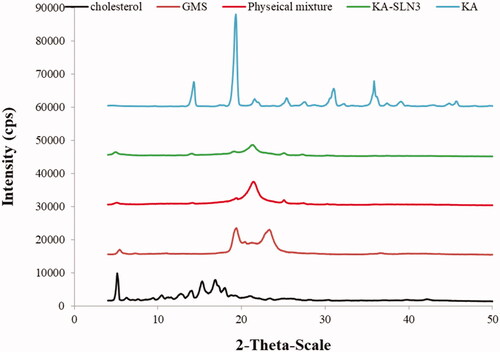
show XRD pattern of KA characterised by distinct sharp peaks at 2θ of 14.204°, 19.2230°, 22.006°, 25.351°, 27.619°, 31.002°, 36.224°, 37.383°, 39.930°. These peaks for KA, Chol and GMS demonstrated the highly crystalline nature of them. The PXRD pattern of physical mixture showed the characteristic peaks at 5.078°, 19.27°, 21.354°, 25.059° and 27.31°. In addition, the XRD pattern of KA-SLN3 appeared the peaks at 2θ: 4.935°, 13.993°, 19.07°, 21.240°, 24°, 25.045°, 27.254°, 30.38° which these peaks did not detect in diffractogram of loaded KA-SLN3 which indicate that KA was well encapsulated within the lipid matrix of SLNs either in molecularly dispersed state and stabilised in amorphous form. In result, XRD, ATR-FTIR and DSC analysis confirmed that there was no significant interaction between drug and other formulation components.
Drug release
The drug release from nanoparticles at a controlled rate can affect drug bioavailability [Citation60]. It has been shown that the use of solid lipids instead of liquid oils in the SLNs can slow down drug mobility compared with a liquid oil and thus allowing for a more sustained/prolonged release of the encapsulated drug. It was reported that the complex factors could be affected the release rate of SLNs, but among the factors, high diffusion coefficient and the large surface area are the dominant factors due to low viscosity or small molecular size in the matrix [Citation61]. If drug is encapsulated into the solid lipid core of SLNs, it is released in a slow and prolonged manner but if drug is loaded in on the matrix surface or the outer shell, it is released in a quick and burst behaviour [Citation62].
In vitro method to characterise diffusion of KA from SLN3 and pure KA for drug release were investigated for 24 h at 37 °C using the paddle method (). The results represented that up to 3.947% ± 0.307 of KA-SLN3 were released over the initial test stages (the first 30 min) followed by prolonged release design. The sustained and slower release rate of KA-SLN3 release after 24 h can be illustrated by complex factor including lower diffusion and dissolution mechanisms of drug from the solid lipid core matrix of the SLNs to dissolution environment.
Figure 5. Dissolution profile of KA solution and KA-SLN3.
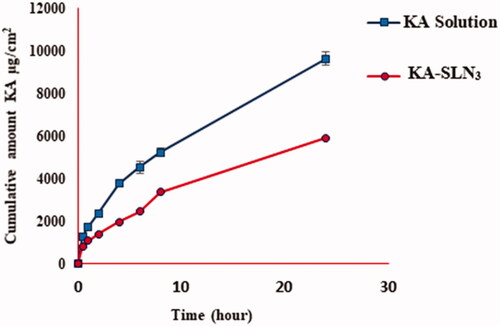
The results showed that the release rate of KA-SLN3 compared with pure KA is the sustained and slower release rate during 24 h. The release results indicated that the SLNs released KA in a controlled behaviour that it could be related to the diffusion of the drug from the solid lipid carrier to the environment according to Baig et al. [Citation63]. In recent years, SLNs open new perspectives in nanomedicines. There have been major efforts to develop SLNs systems that are able to improve the drug release profiles and increase the effectiveness of the formulations for the effective control of drug delivery systems [Citation64–66]. Also, Muller et al. have developed ideas about drug release of SLNs [Citation67–69].
In vitro tyrosinase inhibition activity
Tyrosinase is a cellular target for skin-bleaching preparations and it has been described as an essential metalloenzyme (a copper-containing glycoprotein) and key regulator for melanin biosynthesis. This multifunctional enzyme acts as oxidoreductase and has a main role in catalysing the hydroxylation of monophenols to o-diphenols and the oxidation of o-diphenols to o-quinones. These quinones are highly polymerised with highly speed and high molecular weight pigments called melanin are produced. Excessive synthesis of melanin causes hyperpigmentation disorders [Citation39,Citation70]. Faig et al. reported the effect of KA as a skin-whitening and topical anti-inflammatory agent that has potent tyrosinase inhibition activity on skin pigmentation disorders [Citation71].
KA and KA-SLN3 were screened with L-DOPA (Oxidation of L-DOPA induced tyrosinase activity in mushroom tyrosinase) as the substrate for inhibitory effect on tyrosinase activity investigation. Several concentrations of KA and KA-SLN3 were evaluated (KA-SLN3: 10,00,50,02,50,25,02,00,00,00,000 µg/ml and KA: 1,00,50,25,105 µg/ml) based on this method.
The results showed that KA solution and KA-SLN3 dispersion inhibited the activity of tyrosinase mushroom in a concentration-dependent behaviour. indicates that in KA solution, the maximum enzyme inhibition was 92% at 100 μg/ml and this effect was similar in solutions containing more than 100 µg/ml KA. But these results () show that in KA-SLN3 dispersion the enzyme inhibition increase by increase in nanoparticle concentration. This profile shows a sustained action of enzyme inhibition in KA-SLN3 dispersion between 100–1000 µg/ml concentrations of nanoparticles. The release profile of drug from SLN () can explain this pattern (the maximum percentage of enzyme inhibition was 86% at 1000 μg/ml). Evaluation of these data () indicated enzyme inhibition effect in both solution and SLNs of KA; also a concentration-dependent behaviour and sustained action of enzyme inhibition were observed in KA-SLN3 dispersion due to the slow release of the drug from lipid nanoparticles.
Figure 6. Curves of tyrosinase mushroom activity of KA-SLN3 dispersion and KA solution. (A) The concentrations of KA were 1,00,50,25,105 µg/ml and (B) The concentrations of KA-SLN3 dispersion were 1000, 500, 250, 200,100, 50 and 25 µg/ml. IC50 values were calculated by nonlinear regression using Graph Pad Prism 5.0.
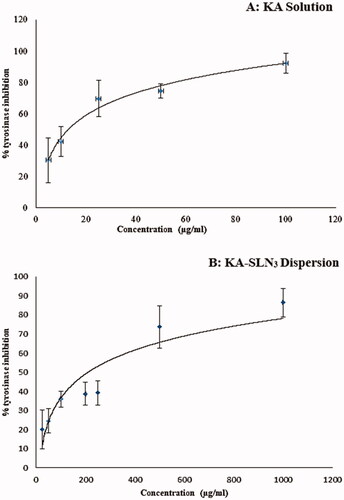
The KA-SLN3 indicated potent inhibitory activity against tyrosinase (IC50 value of 3.84 ± 0.122 μg/ml) as compared with the reference drug KA (IC50 value of 18.31 ± 9.37 μg/ml). Also, based on the results presented above, it was found that KA-SLN3 appears to have more tyrosinase inhibition potency when compared with reference KA solution. These results demonstrated that KA loading into SLNs potentiates the inhibitory activity of KA.
Conclusively the presence of lipid ingredient in the SLN carriers seems to have a capability in the development and efficiency of cosmetic and pharmaceutical preparations due to their easier cellular uptake [Citation22,Citation72].
Ex vivo and in vitro percutaneous absorption study
World Health Organisation (WHO) has the guidelines for membranes obtained from various sources that can be applied for in vitro skin permeation/penetration studies [Citation73]. To evaluate in vitro percutaneous absorption study of a drug, the most relevant skins were human and abdominal rat skin. The use of animal skin such as rat, snake, rodent, monkey and pig skins are reported in the literatures. Studies to evaluate skin permeability have shown that rat skin often has a higher permeability than human skin. however, it is more common than other skin types due to its availability and therefore it can be used during in vitro experiments as candidates to evaluate the transport of drugs across the skin barriers [Citation40].
Human skin compared with the other membranes obtained from various sources has a higher potential for in vitro-in vivo correlations. Human epidermal membrane (HEM) can be prepared from plastic surgery, amputation or autopsies of anatomical regions such as abdomen, breast and legs. However, this skin models could provide a good tool for the quality control of cosmetic preparations [Citation40,Citation74,Citation75].
In this study, drug delivery was compared in KA-SLN3 dispersion against KA solution in similar concentration across HEM, animal and synthesis skins during 24 h. shows ex vivo and in vitro permeation profiles of KA-SLN3 dispersion and KA solution across human, rat and synthetic skins (transdermal delivery) as a function of time. The amount of drug dose permeation of KA-SLN3 dispersion through rat, HEM and synthesis skins (transdermal delivery) were obtained after 24 h permeation, 286.719, 261.575 and 36.835 μg/cm2, respectively. Also, the amount of drug dose permeation of KA solution were 168.629, 156.563 and 21.733 μg/cm2.
Figure 7. Ex vivo and in vitro permeation profiles of KA-SLN3 dispersion and KA solution across human, rat and synthetic skins. Data are presented as mean and standard deviation of three determinations (n = 3).
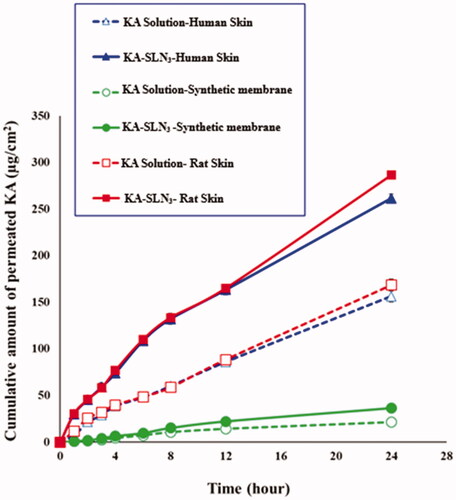
Furthermore, the amount of drug dose permeation across the skins of KA-SLN3 dispersion was significantly more than the KA solution. It looks like that the presence of chemical permeation enhancers such as surfactants and the use of lipid matrix of GMS and Chol in the KA-SLN3 dispersion was the reason for its suitable permeation profile than the KA solution.
Evaluation of these results showed that the permeation of KA-SLN3 dispersion was faster than KA solution (). In many studies, it was reported that the ingredients composing the SLNs have a permeation ability into the deeper skin layers [Citation26,Citation29]. Also, the prolonged release of lipid nanoparticles also contributed to the increase of the permeation of drug [Citation76].
The amount of drug permeated to receptor phase for KA solution was lesser than KA-SLN3 dispersion in all of membranes’ model (p < .05). KA-SLN3 showed more percutaneous absorption across rat skin and HEM (p < .0001). These results were observed via synthetic membrane too (p=.0003).
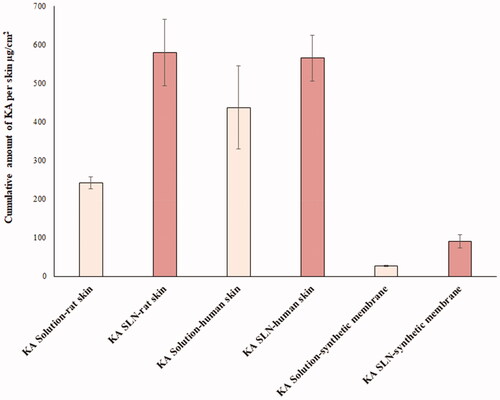
The amount of KA accumulated in different layers of the skin for the optimal KA-SLN3 dispersion was much higher than the KA solution too (). The amount of KA accumulated in human skin was similar in KA solution and KA-SLN3 dispersion (p=.1469). It seems that high deviation between human skin samples was the main reason for this result. The KA-SLN3 dispersion showed more drug retaining in rat skin model in comparison with KA solution (p=.0026). Similar result was observed in synthetic membrane too (p=.003). KA accumulation was similar in rat skin and HEM model (p=.8317).
Figure 8. Cumulative amount of KA in the membrane layers, including rat skin, HEM and synthetic membrane.
These results showed that the highest permeation of drug from KA-SLN3 dispersion was observed for rat skin compared to human and synthetic membranes for 24 h period, which these findings were reported in other studies. Van De Sandt et al. [Citation77] showed the role of skin structure factors such as lipid content, stratum corneum thickness, number of follicles/cm2 and presence or lack of sweat glands in percutaneous absorption via HEM. Many studies of SLNs have been done on the permeability rates in animal and human skins to improve the dermal localisation of various topical therapeutic agents. The permeability rates due to findings are as follows: rabbit > rat > pig > monkey > human. [Citation40,Citation78,Citation79].
These results confirmed the earlier findings about deposition of SLNs in the different layers of skin which can act as a depot system to give sustained delivery of encapsulated drug. SLNs have been shown to improve the dermal localisation of several topical therapeutic agents [Citation80].
Percutaneous absorption study exhibited that the use of KA-SLN3 dispersion can improve the percutaneous accumulation of KA and skin targeting of lipid nanoparticles. Therefore, KA-SLN3 dispersion can help improving the drug levels achieved at the site of disease. Also, in vitro percutaneous permeation studies showed a significant skin targeting effects.
The results of studies done on rat skin showed that KA-SLN3 dispersion can be used to improve topical delivery. More studies also represented that the advantages of using SLNs are promising for the topical delivery of hyperpigmentation, the improvement of topical anti-inflammatory drugs and improving skin disorder therapies (such as aging) [Citation81].
Stability studies
According to , KA-SLN3 were considered as the best formulation in terms of the size, PDI, ZP and EE% with enough colloidal stability. The results of the evaluation of the physical stability studies showed that KA-SLN3 were stable because no significant changes in physical appearance such as precipitate and aggregation of particles were observed, which indicates the good physical stability of SLNs ().
Table 2. Stability information of the KA-SLN3 dispersion after 3 months of storage.
Also, results of KA-SLN3 characteristics () showed that negativity of the ZP was maximum in KA-SLN3 (lipid matrix of GMS and Chol with -27.67 ± 1.89 mV) compared to SA, BW, PA (). For control of colloid stability, nanoparticles must have a large positive or negative ZP [Citation82]. Hence, a ZP of -27.67 ± 1.89 mV for KA-SLN3 was considered significantly high enough to provide colloidal stability to the SLNs. Also, TEM image showed that mixture of span 60 and lipid matrix (GMS and Chol) had produced stable KA-SLN3 dispersion.
Conclusion
In this study, KA-SLN3 dispersion was formulated by using different lipids and optimum formulation selected based on suitable nanoparticle characteristics. The results of the analysis showed that KA was well encapsulated in KA-SLN3 formulation in an amorphous state. These results showed that the KA-SLN3 dispersion can increase the dermal delivery of KA with a higher concentration and controlled release into deeper layers of the skin. Conclusively the presence of lipid ingredient in the KA-SLN3 dispersion carriers seems to have a capability in the development and efficiency of cosmetic and pharmaceutical preparations due to their easier cellular uptake and can be used as a promising and potential novel cosmetic preparation and might open new avenues for treatment of hyperpigmentation disorders.
Acknowledgments
The authors would like to express their sincere and special appreciation to Dr. Mehdi Khalili and their team in Pars Clinic Surgery Centre, Sari, Iran, for their cooperation.
Disclosure statement
Authors declare that there is no conflict of interests in this study.
Additional information
Funding
References
- Sonthalia S, Daulatabad D, Sarkar R. Glutathione as a skin whitening agent: facts, myths, evidence and controversies. Indian J Dermatol Venereol Leprol. 2016;82(3):262–272.
- Hashemi SM, Emami S. Kojic acid-derived tyrosinase inhibitors: synthesis and bioactivity. Pharm Biomed Res. 2015;1(1):1–17.
- Noh J-M, Kwak S-Y, Kim D-H, et al. Kojic acid-tripeptide amide as a new tyrosinase inhibitor. Biopolymers. 2007;88(2):300–307.
- Silpa-Archa N, Kohli I, Chaowattanapanit S, et al. Postinflammatory hyperpigmentation: a comprehensive overview: epidemiology, pathogenesis, clinical presentation, and noninvasive assessment technique. J Am Acad Dermatol. 2017;77(4):591–605.
- James WD, Elston D, Berger T. Andrew’s diseases of the skin e-book: clinical dermatology-expert consult-online and print. Philadelphia: Elsevier Health Sciences, 2011.
- Duarte I, Campos Lage AC. Frequency of dermatoses associated with cosmetics. Contact Derm. 2007;56(4):211–213.
- Nicolaidou E, Katsambas AD. Pigmentation disorders: hyperpigmentation and hypopigmentation. Clin Dermatol. 2014;32(1):66–72.
- Lim JTE. Treatment of melasma using kojic acid in a gel containing hydroquinone and glycolic acid. Dermatologic surgery. 1999;25(4):282–284.
- Bandyopadhyay D. Topical treatment of melasma. Indian J Dermatol. 2009;54(4):303–309.
- Saeedi M, Eslamifar M, Khezri K. Kojic acid applications in cosmetic and pharmaceutical preparations. Biomed Pharmacother. 2019;110:582–593.
- Burnett CL, Bergfeld WF, Belsito DV, et al. Final report of the safety assessment of kojic acid as used in cosmetics. Int J Toxicol. 2010;29(6 Suppl):244S–22473S.
- Serra-Baldrich E, Tribó MJ, Camarasa JG. Allergic contact dermatitis from kojic acid. Contact Derm. 1998;39(2):86–87.
- García-Gavín J. Pigmented contact dermatitis due to kojic acid. A paradoxical side effect of a skin lightener. Contact Dermatitis. 2010;62(1):63–64.
- Fattal E, Bochot A. State of the art and perspectives for the delivery of antisense oligonucleotides and siRNA by polymeric nanocarriers. Int J Pharm. 2008;364(2):237–248.
- Ishihara T, Mizushima T. Techniques for efficient entrapment of pharmaceuticals in biodegradable solid micro/nanoparticles. Expert Opin Drug Deliv. 2010;7(5):565–575.
- Vrignaud S, Benoit J-P, Saulnier P. Strategies for the nanoencapsulation of hydrophilic molecules in polymer-based nanoparticles. Biomaterials. 2011;32(33):8593–8604.
- Khezri K, Saeedi M, Dizaj SM. Application of nanoparticles in percutaneous delivery of active ingredients in cosmetic preparations. Biomed Pharmacother. 2018;106:1499–1505.
- Mishra DK, Dhote V, Bhatnagar P, et al. Engineering solid lipid nanoparticles for improved drug delivery: promises and challenges of translational research. Drug Deliv Transl Res. 2012;2(4):238–253.
- Geszke-Moritz M, Moritz M. Solid lipid nanoparticles as attractive drug vehicles: composition, properties and therapeutic strategies. Mater Sci Eng C Mater Biol Appl. 2016;68:982–994.
- Wang Y-W, Jou C-H, Hung C-C, et al. Cellular fusion and whitening effect of a chitosan derivative coated liposome. Colloids Surf B Biointerfaces. 2012;90:169–176.
- Hussein-Al-Ali SH, El Zowalaty ME, Hussein MZ, et al. Novel kojic acid-polymer-based magnetic nanocomposites for medical applications. Int J Nanomedicine. 2014;9:351–362.
- Ghanbarzadeh S, Hariri R, Kouhsoltani M, et al. Enhanced stability and dermal delivery of hydroquinone using solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2015;136:1004–1010.
- Liu J-J, Nazzal S, Chang T-S, et al. Preparation and characterization of cosmeceutical liposomes loaded with avobenzone and arbutin. J Cosmet Sci. 2013;64(1):9–17.
- Duarah S, Durai RD, Narayanan VB. Nanoparticle-in-gel system for delivery of vitamin C for topical application. Drug Deliv Transl Res. 2017;7(5):750–760.
- Mosallaei N, Mahmoudi A, Ghandehari H, et al. Solid lipid nanoparticles containing 7-ethyl-10-hydroxycamptothecin (SN38): preparation, characterization, in vitro, and in vivo evaluations. Eur J Pharm Biopharm. 2016;104:42–50.
- Akbari J, Saeedi M, Morteza-Semnani K, et al. The design of naproxen solid lipid nanoparticles to target skin layers. Colloids Surf B Biointerfaces. 2016;145:626–633.
- Xu Q, Zhu T, Yi C, et al. Characterization and evaluation of metformin-loaded solid lipid nanoparticles for celluar and mitochondrial uptake. Drug Dev Ind Pharm. 2016;42(5):701–706.
- Luo Y, Teng Z, Li Y, et al. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229.
- Rostamkalaei SS, Akbari J, Saeedi M, et al. Topical gel of Metformin solid lipid nanoparticles: a hopeful promise as a dermal delivery system. Colloids Surf B Biointerfaces. 2019;175:150–157.
- Sharma M, Gupta N, Gupta S. Implications of designing clarithromycin loaded solid lipid nanoparticles on their pharmacokinetics, antibacterial activity and safety. RSC Adv. 2016;6(80):76621–76631.
- Carter CB, Williams DB. Transmission electron microscopy: diffraction, imaging, and spectrometry. Cham: Springer, 2016.
- Kasongo KW, Pardeike J, Müller RH, et al. Selection and characterization of suitable lipid excipients for use in the manufacture of didanosine-loaded solid lipid nanoparticles and nanostructured lipid carriers. J Pharm Sci. 2011;100(12):5185–5196.
- Kumar R, Singh A, Sharma K, et al. Preparation, characterization and in vitro cytotoxicity of Fenofibrate and Nabumetone loaded solid lipid nanoparticles. Mater Sci Eng C Mater Biol Appl. 2020;106:110184.
- Montoto SS, et al. Carbamazepine-loaded solid lipid nanoparticles and nanostructured lipid carriers: physicochemical characterization and in vitro/in vivo evaluation. Colloids and Surfaces B: Biointerfaces. 2018;167:73–81.
- Almeida LN, Araújo TG. Solid lipid nanoparticles: the efficiency carrier for topical delivery of hydrophilic drugs. WJPPS. 2017;6(9):175–189.
- Ghafary S, Ranjbar S, Larijani B, et al. Novel morpholine containing cinnamoyl amides as potent tyrosinase inhibitors. Int JBiolMacromol. 2019;135:978–985.
- Dehghani Z, Khoshneviszadeh M, Khoshneviszadeh M, et al. Veratric acid derivatives containing benzylidene-hydrazine moieties as promising tyrosinase inhibitors and free radical scavengers. Bioorg Med Chem. 2019;27(12):2644–2651.
- Jiménez Z, Kim Y-J, Mathiyalagan R, et al. Assessment of radical scavenging, whitening and moisture retention activities of Panax ginseng berry mediated gold nanoparticles as safe and efficient novel cosmetic material. Artif Cells Nanomed Biotechnol. 2018;46(2):333–340.
- Ashooriha M, Khoshneviszadeh M, Khoshneviszadeh M, et al. 1,2,3-Triazole-based kojic acid analogs as potent tyrosinase inhibitors: design, synthesis and biological evaluation. Bioorg Chem. 2019;82:414–422.
- Beck R, Guterres S, Pohlmann A. Nanocosmetics and nanomedicines: new approaches for skin care. Berlin: Springer, 2011.
- Lin Y-K, Al-Suwayeh SA, Leu Y-L, et al. Squalene-containing nanostructured lipid carriers promote percutaneous absorption and hair follicle targeting of diphencyprone for treating alopecia areata. Pharm Res. 2013;30(2):435–446.
- Kalhapure RS, Mocktar C, Sikwal DR, et al. Ion pairing with linoleic acid simultaneously enhances encapsulation efficiency and antibacterial activity of vancomycin in solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2014;117:303–311.
- Kelidari HR, Saeedi M, Akbari J, et al. Formulation optimization and in vitro skin penetration of spironolactone loaded solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2015;128:473–479.
- Yadav K, et al. Proniosomal gel: a provesicular approach for transdermal drug delivery. Der Pharmacia Lettre. 2010;2(4):189–198.
- Üner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int J Nanomed. 2007;2(3):289.
- Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177.
- Shah R. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J Phys Sci. 2014;25(1):59–74.
- Bazzaz BSF, et al. Antibacterial efficacy of rifampin loaded solid lipid nanoparticles against Staphylococcus epidermidis biofilm. Microb Pathogenesis. 2016;93:137–144.
- Das S, Ng WK, Tan RB. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs?. Eur J Pharm Sci. 2012;47(1):139–151.
- Bhalekar MR, Madgulkar AR, Desale PS, et al. Formulation of piperine solid lipid nanoparticles (SLN) for treatment of rheumatoid arthritis. Drug Dev Ind Pharm. 2017;43(6):1003–1010.
- Liu J, Gong T, Wang C, et al. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: preparation and characterization. Int J Pharm. 2007;340(1-2):153–162.
- Jain A, Sharma G, Thakur K, et al. Beta-carotene-encapsulated solid lipid nanoparticles (BC-SLNs) as promising vehicle for cancer: an investigative assessment. AAPS PharmSciTech. 2019;20(3):100.
- Shenoy VS, Rajyaguru TH, Gude RP, et al. Studies on paclitaxel-loaded glyceryl monostearate nanoparticles. J Microencapsul. 2009;26(6):471–478.
- Kumar R, Yasir M, Saraf SA, et al. Glyceryl monostearate based nanoparticles of mefenamic acid: fabrication and in vitro characterization. Drug Invention Today. 2013;5(3):246–250.
- Butani D, Yewale C, Misra A. Topical Amphotericin B solid lipid nanoparticles: design and development. Colloids Surf B Biointerfaces. 2016;139:17–24.
- Rompicharla SVK, Bhatt H, Shah A, et al. Formulation optimization, characterization, and evaluation of in vitro cytotoxic potential of curcumin loaded solid lipid nanoparticles for improved anticancer activity. Chem Phys Lipids. 2017;208:10–18.
- Gardouh AR. Design and characterization of glyceryl monostearate solid lipid nanoparticles prepared by high shear homogenization. BJPR. 2013;3(3):326–346.
- Freitas C, Müller R. Correlation between long-term stability of solid lipid nanoparticles (SLN™) and crystallinity of the lipid phase. Eur J Pharm Biopharm. 1999;47(2):125–132.
- Kumar VV, Chandrasekar D, Ramakrishna S, et al. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335(1-2):167–175.
- Pancholi K, Stride E, Edirisinghe M. In vitro method to characterize diffusion of dye from polymeric particles: a model for drug release. Langmuir. 2009;25(17):10007–10013.
- Righeschi C, Bergonzi MC, Isacchi B, et al. Enhanced curcumin permeability by SLN formulation: the PAMPA approach. LWT-Food Science and Technology. 2016;66:475–483.
- Reddy LH, Murthy R. Etoposide-loaded nanoparticles made from glyceride lipids: formulation, characterization, in vitro drug release, and stability evaluation. AAPS PharmSciTech. 2005;6(2):E158–E166.
- Baig MS, Ahad A, Aslam M, et al. Application of Box-Behnken design for preparation of levofloxacin-loaded stearic acid solid lipid nanoparticles for ocular delivery: optimization, in vitro release, ocular tolerance, and antibacterial activity . Int J Biol Macromol. 2016;85:258–270.
- Attama AA, Umeyor CE. The use of solid lipid nanoparticles for sustained drug release. Ther Deliv. 2015;6(6):669–684.
- Devi R, Jain A, Hurkat P, et al. Dual drug delivery using lactic acid conjugated SLN for effective management of neurocysticercosis. Pharm Res. 2015;32(10):3137–3148.
- Pyo SM, Meinke M, Klein AF, et al. A novel concept for the treatment of couperosis based on nanocrystals in combination with solid lipid nanoparticles (SLN). Int J Pharm. 2016;510(1):9–16.
- Müller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54:S131–S155.
- Üner M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): their benefits as colloidal drug carrier systems. Die pharmazie Int J Pharm Sci. 2006;61(5):375–386.
- Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95(3):627–638.
- Emami S, Hosseinimehr SJ, Shahrbandi K, et al. Synthesis and evaluation of 2(3H)-thiazole thiones as tyrosinase inhibitors. Arch Pharm (Weinheim). 2012;345(8):629–637.
- Faig JJ, Moretti A, Joseph LB, et al. Biodegradable kojic acid-based polymers: controlled delivery of bioactives for melanogenesis inhibition. Biomacromolecules. 2017;18(2):363–373.
- Shrotriya S, Ranpise N, Satpute P, et al. Skin targeting of curcumin solid lipid nanoparticles-engrossed topical gel for the treatment of pigmentation and irritant contact dermatitis. Artif Cells Nanomed Biotechnol. 2018;46(7):1471–1482.
- Byford T. Environmental health criteria 235: dermal absorption. Int J Environ Stud. 2009;66(5):662–663.
- Huong SP, Bun H, Fourneron J-D, et al. Use of various models for in vitro percutaneous absorption studies of ultraviolet filters. Skin Res Technol. 2009;15(3):253–261.
- SCCS. Basic criteria for the in vitro assessment of dermal absorption of cosmetic ingredients. Brussels: European Commission Health & Consumers, 2010.
- Wang X-R, Gao S-Q, Niu X-Q, et al. Capsaicin-loaded nanolipoidal carriers for topical application: design, characterization, and in vitro/in vivo evaluation. Int J Nanomedicine. 2017;12:3881–3898.
- van de Sandt JJ, Meuling WJ, Elliott GR, et al. Comparative in vitro-in vivo percutaneous absorption of the pesticide propoxur. Toxicol Sci. 2000;58(1):15–22.
- Escribano E, Obach M, Arévalo MI, et al. Rapid human skin permeation and topical anaesthetic activity of a new amethocaine microemulsion. Skin Pharmacol Physiol. 2005;18(6):294–300.
- Liu J, Hu W, Chen H, et al. Isotretinoin-loaded solid lipid nanoparticles with skin targeting for topical delivery. Int J Pharm. 2007;328(2):191–195.
- Borgia SL, et al. Lipid nanoparticles for skin penetration enhancement—correlation to drug localization within the particle matrix as determined by fluorescence and parelectric spectroscopy. J Control Release. 2005;110(1):151–163.
- Garcês A, Amaral MH, Sousa Lobo JM, et al. Formulations based on solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for cutaneous use: a review. Eur J Pharm Sci. 2018;112:159–167.
- Riddick T. Control of colloid stability. Zeta Potential. Staunton, VA: Zeta-Meter Inc, 2007. p. 198–200.