
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Tumour development is not only an independent event of genetic mutation and overgrowth of tumour cells but is the result of a synergistic interaction between a malignant tumour and its surrounding tumour stromal microenvironment. In this paper, we address the shortcomings of current tumour therapy by focussing on the tumour itself and the surrounding microenvironment to achieve a two-pronged targeting model. In this paper, a dual-targeting, pH/reactive oxygen species (ROS) sensitive nano-drug delivery system for tumour cells and CAFs was designed. A hyaluronic acid (HA) with CD44 receptor targeting on the surface of tumour cells was selected as the main carrier material, and a dipeptide Z-glycine-proline (ZGP) with specific targeting of fibroblast activating protein (FAP) on the surface of CAFs was modified on HA to achieve precise targeting of CAFs, open the physical barrier of tumour cells and improve the deep penetration effect of the tumour, while introducing thioketone bond and ketone condensation bond to take advantage of the highly reactive ROS and low pH microenvironment at the tumour site to achieve chemical bond breaking of nano micelles encapsulating paclitaxel (PTX), drug release, and thus drug aggregation at the tumour site and improved bioavailability of the drug.
Introduction
Malignancies are a growing global public health problem. According to the 2015 annual report of the International Health Organisation, liver cancer, as the fifth most prevalent malignancy in the world, ranks second in the global malignancy mortality rate [Citation1]. Survey data show that the incidence of liver cancer is mainly concentrated in developing countries, with the incidence in China being particularly serious [Citation2]. Liver cancer is characterised by being difficult to detect, difficult to treat and progresses rapidly. Currently, the main treatments include surgical resection, radiotherapy, ablation therapy and interventional therapy. Although these treatments will also kill some of the peri-cancerous tissues, it is difficult to inhibit tumour recurrence from the root cause. Therefore, more and more researchers are focussing on two-pronged targeted treatment modalities for the tumour itself and the surrounding microenvironment.
More and more research demonstrated that fibrous TME played an important role in accelerating tumour progression and deterioration. Since the rare lethal tumour cells "seeds" are cultivated and protected by the rich tumour matrix "soil", targeted nano-drug therapy based on these complex features of cancer cells cannot fully control the pathological processes of cancer development by TME. Moreover, after passing through the tumour blood vessels, the nano-drugs targeted by dense ECM, cancer cells are often retained in the tumour matrix and unable to penetrate inside the tumour, resulting in an insufficient concentration of local drug in the deep tumour to inhibit the deteriorating [Citation3,Citation4] of the tumour cells located in the deep part. Therefore, exerting appropriate TME remodelling, reversing the tumour-promoting role, and improving deep drug penetration were key to effectively improving cancer nano treatment.
Epidemiological and clinical studies have shown that tissue fibrosis in certain organs such as the liver and pancreas is a precursor to corresponding cancer [Citation5–7]. For example, stationary pancreatic and hepatic stellate cells can obtain CAFs-like phenotypes in pancreatic and liver cancer. CAFs, as a network cell, are steady with little antigen misfortune and treatment obstruction; enormous aggregate contrasts of tumour cells between people, while grid cells are non-harmful cells with somewhat single phenotype. The fibroblast-activated protein (FAP) is one of the most important molecular markers on the CAFs surface, which became a potential target for tumour immunotherapy [Citation8–10]. FAP is a type II complete membrane serine protease overexpressed by CAFs, selectively expressed in over 90% of human epithelial tumours, and is considered a generic tumour antigen and a promising target for CAFs depletion [Citation11,Citation12]. Z-glycine-proline (ZGP) is a small molecule peptide that can specifically target FAP [Citation13–15].
Natural polysaccharides, with the attributes of good biocompatibility, degradability, easily soluble in water, low toxicity, and easy modification, have been more and more favoured by researchers and widely used in the research of nano-drug delivery systems [Citation16–18]. As of now broadly utilised are hyaluronic corrosive (hyaluronic corrosive, HA), (chitosan, CS), Columbus polysaccharide, lentinan polysaccharide, angelica polysaccharide, and so forth [Citation19–22]. Natural polysaccharides as drug carrier, can not only improve the stability of drugs, reduce the toxic side effects of drugs, avoid accumulation and residue in normal tissues, but also has some biological characteristics, such as antioxidant, adjust the body’s immune functions [Citation23,Citation24]. The CD44 receptor is an adhesion receptor present on the surface of most tumour cells and is also highly expressed on macrophages and fibroblasts at inflammatory sites in the meantime [Citation25–27]. HA can be explicitly adsorbed with CD44 receptors as target particles for CD44 receptors, and hence HA is broadly utilised in the development of nanocarriers.
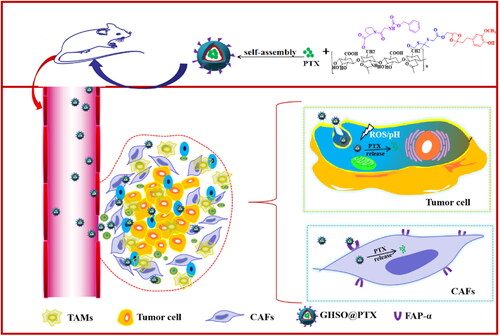
Herein, given the current disadvantages of tumour treatment, this paper focussed on the tumour itself and its surrounding microenvironment designs a dual-pronged targeted treatment mode and hoped to achieve a better tumour treatment effect. First, hyaluronic acid (HA) that targets the CD44 receptor on the tumour cell surface was selected as the main backbone of the biophilic vector material, and introducing z-glycine-proline (ZGP) on its surface can specifically target the FAP receptor on the CAFs surface, achieving the purpose of double targeting tumour cells and CAFs. Then, sensitive groups (thiones and acetones) responding to high ROS and low pH in tumour cells were introduced to achieve drug release in tumour cells, reducing damage to other non-tumour cells. The nano-micelles as the drug delivery system improved the solubility of PTX and the stability of the drug. When the nano-micelles GHSO@PTX reached the tumour site, it first made contact with the matrix cells around the tumour cells, killing it by the endocytic release drug through specific binding to CAFs. CAFs were killed, both opened the barrier and reduced TGF-β secretion, reducing the promoting effect on tumour cells and reduce the degree of fibrosis. The whole mechanism () realised a targeted treatment strategy of killing tumour cells and inhibiting the tumour-promoting microenvironment.
Figure 1. Schematic illustration of the structure of GHSO@PTX.
Materials and methods
Chemicals, cell lines, and Animals
Zingiberone, ZGP and PTX were obtained from Aladdin Chemistry Co., Ltd. TKL was received from Shanghai Chuangyan Chemical Technology Co., Ltd. HA was achieved from Huaxi Forida Biotechnology Limited. N-(3-Dlmethylamlnopropyl)-N’-ethylcarbodllmide hydrochloride (EDC), Dimethyl Sulphoxide (DMSO), 4-Dimethylaminopyridine (DMAP), 1-Hydroxybenzotriazole Hydrate (HOBT), Succinimide (NHS) were bought from Tianjin Yongda Chemical Reagents Co., Ltd. H2O2 was gained from Aladdin reagent net.3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO), DMEM (High Glucose), and foetal bovine serum (FBS) were purchased from Junshuo Biotech Co., Ltd (Yantai, China).
NIH 3T3 (Normal tissue fibroblasts) was got from BeNa Culture Collection (Beijing, China). The SMMC-7721 cell line (hepatoma carcinoma cell) was obtained from the Shandong Academy of Pharmaceutical Science.
The NU/NU female nude mice were procured from Beijing Vital River Laboratory Animal Technology Co. Ltd (SPF grade, 6–8 weeks, 18–22 g). The experimental process was carried out in strict accordance with the relevant regulations of the Committee on the Management and Use of Experimental Animals in Yantai University and the National Institute of Health Guide for the Care and Use of Laboratory Animals. Nude mice were divided into six groups with four mice in each group. These groups were: Free PTX group, HSO@PTX(-) group, HSO@PTX(+) group, GHSO@PTX(-) group, and GHSO@PTX(+) group.
Synthesis and characterisation of GHSO
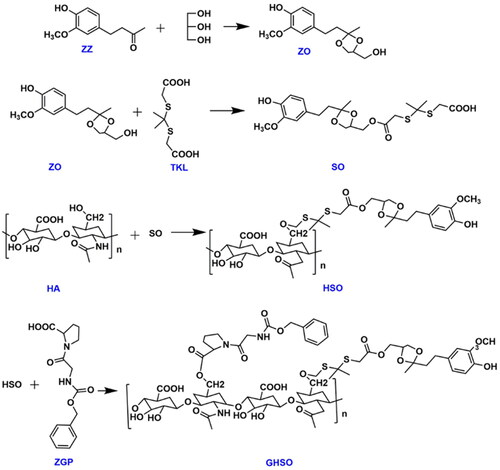
In three bottles of 150 ml with a thermometer, water distributor, reflux condensate tube, added zingiberone: glycerol: cyclohexane (molar ratio of 1: 1.0 ∼ 1.5: 1), added p-toluene sulphonic acid as catalyst, the mole ratio of p-toluene sulphonic acid and gingerone was 1: 150 ∼ 250, this reaction condensed reflux 6 h at 92 °C. After the removal of toluene after the reaction, ethyl acetate and saturated sodium carbonate were added for washing extraction 3 ∼ 4 times and 45 ml/times and dried. Purpurified compounds were separated after drying, when the oil ether: ethyl acetate =1: 1, the products were collected at this proportion, and ZO compounds were made.
The formamide (6 ml), ZO (100 mg), TKL (27.86 mg), EDC, and DMAP were added into the 50 ml round bottom flask and magnetically stirred until they were completely dissolved at room temperature. After stirring the reaction at room temperature for half an hour, the reaction temperature was gradually raised to 40 °C in an oil bath. After 24 h the product ginger azone-thiazone (so) was obtained. Further, 150 mg of HA was added into the reaction system, and the crude product was continuously reacted for 48 h. The crude product was dialysed by using a dialysis bag (2000 Da MWCO). HA-SS-SO(HSO) was obtained of continuous drying using a freeze dryer. Finally, The formamide (7 ml), HSO (92.6 mg), ZGP (20 mg), EDC, and NHS were added into the round bottom flask and magnetically stirred until they were completely dissolved. After a reaction of 24 h, at 30 °C, they were transferred to the molecular weight dialysis bag of 2000 Da under the above dialysis method and freeze-dried to obtain the final carrier material GHSO. The synthetic route of this reaction is shown in .
Figure 2. Design route of GHSO.
1H-NMR spectroscopy was used to detect the chemical structure of GHSO. A total of 10 mg of GHSO was weighed and dissolved in 0.6 ml of D2O and d6-DMSO to detect the chemical shifts.
Preparation of the GHSO@PTX nano-micelles
The synthetic GHSO can load PTX by self-assembly. GHSO (10 mg) and PTX (1 mg) dissolved in formamide were added into the dialysis bag (2000 Da). The dialysis water was replaced almost every 2 h till the formamide in the dialysis bag was completely gone. Thereafter, we obtained the nano-micelles of GHSO@PTX. IR spectra for GHSO materials were tested and determined at room temperature.
Characterisation of the GHSO@PTX nano-micelles
The HSO@PTX and GHSO@PTX micelles obtained by the above preparation method were determined by the particle size, PDI, potential. The nano-micelles HSO@PTX and GHSO@PTX, obtained by the above preparation method were repeatedly dropped on the copper network, naturally dried with phosphotungstic acid, and the morphology of the nano-gels was observed with a transmission radio microscope. The Encapsulation efficiency (EE%) and Drug loading (DL%) capacity of nano-micelles HSO@PTX and GHSO@PTX were determined by HPLC.
Experimental study on in vitro release of the GHSO@PTX nano-micelles
To examine the responsive release characteristic of PTX from GHSO@PTX, the release profiles were examined by using the dialysis method. First, we examined the ROS-responsive PTX release capacity of the GHSO@PTX. Briefly, 1 ml GHSO@PTX was placed in a dialysis bag (MWCO 2000 Da) and immersed in 50 ml PBS buffer containing 0.5% (v:v) Tween 80 with different H2O2 and pH concentrations (pH 7.4, H2O2 0.1 mM; pH 7.4, H2O2 1 mM; pH 7.4, H2O2 10 mM; pH 5.8, H2O2 0.1 mM; pH 5.8, H2O2 1 mM; pH 5.8, H2O2 10 mM). The whole system was agitated at 37 °C and sampled at pre-defined time points. The sample solution was taken from different time points and different release media, after the filter film exceeded 0.22 μm, the concentration content of PTX was determined with the high-performance liquid phase and the cumulative release amount under different release media.
Cytotoxicity assessment
To investigate the safety of the blank carrier material, the SMMC-7721 cells or CAFs were co-incubated with blank GHSO of various doses (10–500 μg/mL) at 37 °C. 20 μL of MTT (5 mg/mL) was added to the per well. After 4 h, the supernatant medium was taken the place of DMSO (200 μL) and shaken for 10 min. The microplate reader (Thermo Fisher Scientific Co., Waltham, MA) was used to determine the absorbance.
The effects of PTX preparations on SMMC-7721 cells and CAFs in vitro were examined by using an MTT assay. To compare the viability effects, Free PTX, HSO@PTX, and GHSO@PTX were fully dissolved in DMEM for final PTX concentrations from 1.25 μg/mL to 40 μg/mL. Typically, after counting, SMMC-7721 cells or CAFs cells were uniformly dispersed in cell culture medium at a density of 10,000 cells/mL, and 200 μL of the cell suspension was placed in each well of a 96-well plate, and cultured overnight until the cells were fully adherence. Subsequently, fresh DMEM-containing PTX preparations were replaced and incubated for an additional 24 or 48 h. The microplate reader was conducted to gauge the relative cell viability. We could calculate cell survival (%) according to the following formula:
Cellular uptake
The fresh medium containing Free PTX, HSO@PTX micelles, or GHSO@PTX micelles were added to replace the original medium after 24 h (48 h), respectively. The SMMC-7721 cells or CAFs were then cultured for 0.5 h, 1 h, 2 h, and 4 h (PTX concentration: 20 μg/mL) or for 4 h with the following different concentrations of PpTX: 5 μg/mL, 10 μg/mL, 20 μg/mL, and 40 μg/mL in the above atmosphere. The consequence of cellular uptake was observed by an inverted fluorescence microscope to have a qualitative analysis.
In vitro penetration of different nanoparticles in tumour cells and fibroblast-mixed multicellular hybrid tumour spheroids
To mimic the solid tumour microenvironment in which tumour cells and fibroblasts grow together, we established CAFs cells & SMMC-7721 cells cocultured hybrid tumour spheroids. The in vitro tumour penetration ability of Cur-labeled nanoparticles with or without ZGP incubation was investigated on hybrid tumour spheroids. The multicellular tumour spheroids were established according to a previous report.28 Briefly, 80 μL of hot 2.0% agarose solution (w/v) was added to 96-well plates and then cooled to room temperature. SMMC-7721 cells were mixed with fibroblasts at a ratio of 2:1, seeded in 96-well plates, and cultured for 2–3 days to grow into a spheroid. Then, HSO@Cur and GHSO@Cur were added for incubation to observe the effect of penetration.
Construction of animal tumour models
The nude mice are housed in a sterile environment, the shavings and sterilised water are changed regularly and fed promptly to ensure. The nude mice are kept in a clean environment. When the nude mice reach a certain size of about 20 g, they are inoculated with axillary tumours. The number of inoculated cells was 2 × 106. After the tumour had reached a certain size, all nude mice were randomly divided into 3 groups according to the principle of random allocation. All nude mice were randomly divided into 3 groups according to the principle of random allocation, with 3 mice in each group, namely Free DiR group, HSO@DiR group and GHSO@DiR group.
In vivo distribution of mice xenografted
The in vivo distribution was measured by In-Vivo FX Pro in vivo imaging system. To observe the accumulation of PTX, HSO@PTX micelles, and GHSO@PTX micelles in vivo, the Free DiR, HSO@DiR, and GHSO@DiR micelles (at the DiR concentration at 500 μg/mL) were injected into SMMC-7721 and CAFs tumour-bearing mice via the tail vein. On hour 12 after injection of kinds of DiR, and the tumour and the major organs of mice were collected and the fluorescence intensity of various organs was monitored in vitro by In-Vivo FX Pro.
In vivo pharmacodynamics study
Docking nude mice were randomly divided into 6 groups, 3 in each group, raised separately and marked, and began to give the medicine when the tumour grew to the appropriate volume. The administration group was Saline, Free PTX, HSO@PTX(-), HSO@PTX(+), GHSO@PTX(-), GHSO@PTX(+), with high and low concentrations of the high concentration. The tumour grew up to 100 mm3 for tail IV administration (5 mg/Kg), administered every 3 days. Each administered, weight and tumour volume were measured for 30 days. Three days after the administration, all the naked rats were euthanized, and the heart, liver, spleen, lung, kidney, and tumour tissue were dissected and stored in 4% polyformaldehyde respectively. The weight change curve, volume change and tumour suppression rate of each group were drawn according to the experimental records. The specific formula is as follows:
Where L and W are the length and width of the tumour, respectively; Wcontrol is the tumour weight of the control group and Wtested is the tumour weight of the experimental group.
Preliminary histological study
Remove the isolated tissue previously fixed to polyformaldehyde, bury paraffin, and dry in a 60 °C oven after slicing. After that, the isolated organ tissue sections and tumour tissue of different drug administration groups were stained by H&E, sealed after dyeing the volatile xylene, observed under the microscope, and took photos.
Immunohistochemistry
To evaluate the growth inhibition of the tumour cells, the expression of Ki 67 in the tumour cells was investigated. To verify the inhibitory effect of GHSO@PTX on the tumour microenvironment and antifibrosis, α-SMA and Masson staining were performed, and the number of α-SMA-positive cells and collagen content in the tumour tissues were examined.
Statistical analyses
All data are performed with Student’s t-test and ANOVA. The quantitative data were presented as mean ± standard deviation (SD).
Results and discussions
Characterisation of GHSO materials
1H-NMR
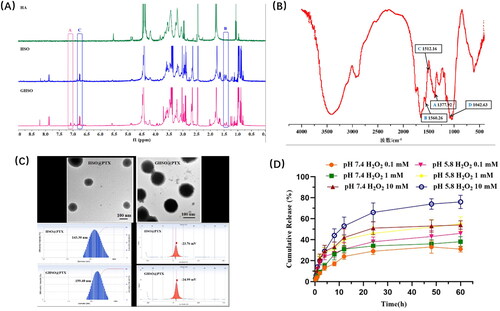
The 1H-NMR spectroscopy of HA, HSO, and GHSO was clearly shown in . The methyl peak for TKL was observed at about δ: 1.5. 1H-NMR spectra revealed an absorption peak at about δ: 4.77, which was -OH in ZO. The appearance of the signal peak at δ: 7.1 verified the presence of ZGP, indicating that the new product of GHSO had been synthesised successfully.
Figure 3. (A) The 1H-NMR spectra of HA, HSO and GHSO. (B) The FT-IR spectra of GHSO. (C) The TEM morphology, size, zeta of HSO@PTX micelles and GHSO@PTX micelles. (D) Curve of GHSO@PTX releasing PTX in different environments.
FT-IR
FT-IR spectra of GHSO materials were shown in . A referred to together dimethyl group, coupled to split into two special shape spectral bands near 1375 cm-1, The double-peak in the picture testified the connection of the TKL. B, C referred to the skeleton vibration peak of the aromatic ring, D referred to the vibration coupling peak of ZO, 1083 cm-1.
Particle size, zeta potential, morphology, EE%, and DL%
The results of showed that the particle size of HSO@PTX micelles was 143.30 ± 17.00 nm, the PDI was 0.236 ± 0.004, the potential was −23.76 ± 5.12 mV, the encapsulation efficiency (EE%) was 46.47 ± 2.80%, and the drug loading (DL%) was 4.57 ± 0.68%. The particle size of GHSO@PTX micelles was 159.40 ± 14.30 nm, PDI was 0.159 ± 0.06, the potential was −24.99 ± 4.73 mV, EE% was 49.61 ± 3.52% and DL% was 4.72 ± 0.39%. The electron microscopy results show that the two micelles were relatively uniform in size and spherical in shape.
Drug release investigation
GHSO@PTX at different H2O2 concentrations and pH conditions are shown in below. It can be seen from the figure that each group was slowly released under the conditions and had no sudden release. For the first three groups, the control variable pH remained unchanged at 7.4. As we can see that the H2O2 concentration increased from 0.1 mM to 1 mM, to 10 mM, which was 31%, 38%, and 34% respectively. See that when the H2O2 concentration, the cumulative release increased significantly at 10 mM, for fracture of affinity vector material GHSO under high concentration H2O2, promoting drug release. When the H2O2 concentration was constant and all 10 mM, it showed obvious growth in pH 5.8(76%) compared with pH 7.4(54%), which proved a promoting effect of low pH on nano-micelles. The analysis reason was pH 5.8, ginger acetone bond fracture of GHSO, the conformational change of two affinity carrier materials, micelles dissociation, and promote drug release.
Cytotoxicity assessment
The cytotoxicity results of nano-micelles to SMMC-7721 cells and CAFs were shown in . represented the growth inhibition of blank micelles on both cells at 24 h. It was obvious from the figure that the toxicity of blank micelles is very small, with a survival of 85% at the concentration of 500 μg/mL, demonstrating that the in vivo safety of our designed affinity vector material GHSO was good.
Figure 4. The cytotoxicity of different formulations 24 h (A) and 48 h (B) to SMMC-7721 cells; the cytotoxicity of different formulations 24 h (C) and 48 (D) to CAFs cells; (E) The cytotoxicity of blank micelles to SMMC-7721 and CAFs cells. (*p < .05).
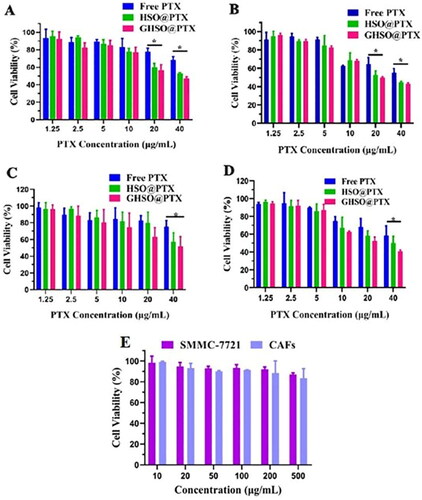
represented cytotoxicity results on SMMC-7721 cells by different administration groups at 24 h and 48 h, respectively. Meanwhile, represented cytotoxicity results on CAFs cells by different administration groups at 24 h and 48 h, respectively. The results showed that with the increase of drug concentration, the survival rate of each drug group decreased to different degrees. At the same drug concentration, the cell survival of the nano-micelles group was lower than that of Free PTX, indicating that the nano-micelles group had a stronger lethal effect on tumour cells, probably because HA played the main role in the targeting of tumour cells SMMC-7721. In , the GHSO@PTX group showed the strongest cytotoxicity, possibly because it could target CAFs, to enhance the therapeutic effect of tumours.
Cellular uptake
represented the uptake effect of Free Cur, HSO@Cur, GHSO@Cur on SMMC-7721 cells (A) and CAFs cells (B) at different times. The results in the figure showed that the intake of both cells in each administration group has increased over time. In , the nano-micelles group uptake effect was much better than in the Free drug group. In ), the uptake effect of the nano-micelles bunch was superior to the Free medication bunch simultaneously, and the take-up impact of the nano-micelles GHSO@Cur bunch was ideal, additionally because of its designated impact on CAFs.
Figure 5. (A) The uptake of SMMC-7721 (A) and CAFs (B) after different administration times; The uptake of SMMC-7721 (C) and CAFs (D) after different administration concentration. (B) The penetration of GHSO@Cur (A) and HSO@Cur (B) in a 3D tumour sphere. C. The fluorescence of nude mice at different time intervals (2 h, 4 h, 8 h, 12 h and 24 h); The fluorescence intensity images of isolate organ and tumour tissues in nude mice.
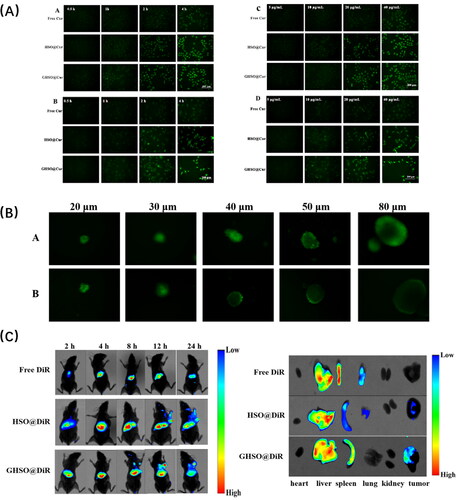
) represented the uptake effect at different drug concentrations. It could be seen from the figure that, as the drug concentration increases, the intake effect of each drug administration group has increased to varying degrees. In ), the uptake of HSO@Cur and GHSO@Cur was better compared with the Free Cur group, proving the superiority of micelles. Another reason was that HA could target the CD44 receptor on the surface of tumour cells, but the two nano-micelles were not significantly different because ZGP was specific for targeting the FAPα enzyme on CAFs cell surface and does not promote the uptake of tumour cells. In Figure5(A,D), the nano-micelles group was also better than the free drug group, but the GHSO@Cur group was significantly stronger than the HSO@Cur group because GHSO is specific for targeting CAFs, and thus more uptake superiority, consistent with the initial scenario.
In vitro penetration of multicellular hybrid tumour spheroids
In this experiment, Cur was selected as a fluorescent substance, loaded in the two affinity vector materials HSO and GHSO, to investigate the in vitro tumour ball penetration effect of two nano-micelles. represented the GHSO@Cur group, Figure5(B) represented the HSO@Cur group, and the middle black site represented the site where the nano-micelles failed to reach. It was obvious that in the 3D tumour ball of different particle sizes, the deep penetration effect of the GHSO@Cur group was significantly stronger than that of the HSO@Cur group. For the analysis reason, it might be that the double-targeted nano-micelles GHSO@Cur group could first target the CAFs, to open the barrier and better achieve the deep penetration of the nano-micelles.
In vivo fluorescence imaging
The results in show that the HSO@DiR and GHSO@DiR groups compared with the control Free DiR solution group, DiR can be better delivered to the tumour tissue at about 8 h and accumulate more to the tumour tissue site over time. It was worth noting that the GHSO@DiR group reached the tumour tissue one step earlier than the HSO@DiR group and had a stronger fluorescence intensity at the tumour. At the early stage of administration, the nano-micelles group was distributed to the main organs. Over time, the impact on the main organs had decreased a lot. By 24 h, the double-targeted GHSO@DiR group had the least impact on the main organs but had the most savings in the tumour site.
The figure on the right represents the results of fluorescence imaging of isolated tissue and organs after administration of 12 h. The results showed that there was no accumulation of the tumour site in the control Free DiR solution group, and both groups of nano-micelles could deliver DiR to the tumour site, demonstrating the superiority of the nano-drug delivery system, and the targeting of HA to tumour cells achieved DiR delivery in the HSO@DiR group. The dual-targeted GHSO@DiR group had the most savings in the tumour site, possibly due to the ZGP targeting of CAFs promoting nanobelts into the tumour tissue. The accumulation of three groups of organs was mainly the liver and spleen, the analysis reason might be because the liver as an important metabolic organ of the body, these foreign substances were metabolised by the liver, leading to the accumulation in the liver site; or because preparations are swallowed by macrophages in the body’s mesh endothelial cell system, with more macrophages in the liver and spleen, leading in the most accumulation of the liver and spleen parts.
In vivo pharmacodynamics study
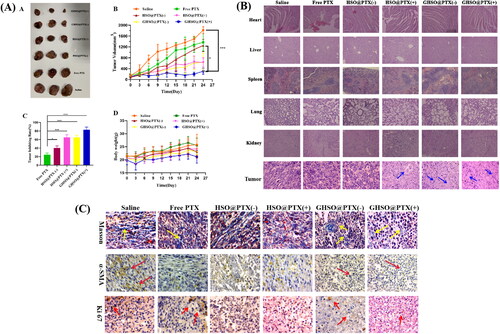
Both ) could intuitively observe that the HSO@PTX and GHSO@PTX groups are significantly better than the free drug PTX groups, possibly because the nano-drug delivery system could better deliver the drug to the tumour site, improve the effective content of the drug in the tumour site, and play a good therapeutic role, demonstrating the characteristics of pH sensitivity and ROS response. GHSO@PTX group compared with the HSO@PTX group, the treatment effect was better, the analysis might be because GHSO@PTX could specifically target the matrix CAFs, around tumour cells for nanoparticles removal obstacles, promote nanoparticles to better achieve deeper penetration of tumours. The treatment effect of the high concentration of GHSO@PTX(+) group was higher than that of GHSO@PTX(-), and there was no naked rat death caused by a high concentration during administration, which also affirmed the good compatibility of the carrier material, improved the treatment concentration of the drug and reduced the toxic side effects of the drug. As can be seen from , the weight of naked rats in the nanoparticles group did not change significantly in the early administration and showed a slight decrease in the later administration, which proved that the nanoparticles were less toxic and have certain safety in the body.
Figure 6. (A) The photographs of tumours from different groups. (B) The tumour volume changes from different groups. (C) The tumour inhibition rate from different groups. (D) The body change of tumour-bearing nude mice. B.H&E staining of major organs and tumour issue in different administration groups. (C) Masson staining, α-SMA staining and Ki 67 expression of tumour tissue. n = 3; *p < .05, **p < .01.
Preliminary histological study
It could be seen from that HSO@PTX and GHSO@PTX had very small damage to the heart, liver, spleen, lung, and kidney of naked rats, combined with the weight change of naked rats, but also more proved the safety of the two affinity carrier materials, reduced the toxicity of drugs, and played a good transport role. It could be seen from the H&E staining results of the tumour tissue of nude tumour rats, there was no damage in the tumour tissue of the saline group, which as a control, observed the free drug group and no excessive damage to the tumour tissue. The analysis might be that the free drug PTX failed to reach the tumour tissue too much and did not play an effective therapeutic effect. Observe the preparation group, we found obvious damage to tumour tissue and large death of tumour cells, which proved the inhibitory effect of a particle on tumour cells, especially the high concentration GHSO@PTX(+) group, the loosest distribution, and obvious core consolidation shrinkage, and proved the best treatment effect of double-targeted preparation high concentration GHSO@PTX(+) group.
Immunohistochemistry
Ki 67 is an important marker of cell proliferative activity. High-level expression of Ki 67 represents a strong cell proliferative activity and can be used as a reliable evaluation indicator after tumour treatment. The results showed that the expression of Ki 67 in the high concentration of GHSO@PTX(+) group was significantly reduced, proving that the tumour cell proliferation activity was significantly inhibited and that the double-targeted nanoparticles GHSO@PTX had a good role in tumour therapy. CAFs cells are α-SMA positive cells, α-SMA staining can brown α-SMA positive cells, and we can judge using the number of brown cells. It can be seen from that compared with the saline group and the free drug group, the significant reduction of brown cells in the GHSO@PTX group was reduced, indicating that the number of CAFs cells was significantly reduced. The reason analysis may be because GHSO can target the FAP α enzyme on the CAFs surface, and then enter CAFs, to realise the lethal effect on CAFs, thus reducing the promoting effect of the tumour microenvironment on the growth of tumour cells. Masson tricolour staining is a classic staining method for collagen cellulose, which dyes collagen cellulose blue. Collagen cellulose is secreted by CAFs cells, deposited in microenvironments, leading to liver fibrosis and promoting the development of tumour cells. From the figure, the blue lines of Preparation group GHSO@PTX were significantly reduced, indicating decreased collagen secretion, also proving that CAFs are inhibited, while decreased collagen deposition, but also indicating inhibiting the development of liver fibrosis.
Conclusions
In chemical synthesis, the affinity nano-carrier material HSO, with pH sensitivity and ROS response was synthesised to connect the deep penetration of the target with CAFs targeting function, synthesise the final carrier material GHSO, and conduct a series of characterisation of the carrier material to verify the successful synthesis. Nanomicelles were prepared by the carrier load drug PTX, dialysis, and investigated the particle size, potential, and electric mirror. In vitro release experiments verified the pH sensitivity and ROS responsiveness of the carrier material. In the cell experiment, the 3D tumour ball deep penetration experiment was studied to investigate the cell penetration of the in vitro tumour ball through multiple cell cultures in vitro. In the immunohistochemical experiment, the obvious reduction of Ki 67 meant that the proliferation activity of tumour cells was suppressed and the decrease of α-SMA-positive cells, which proved the targeting of GHSO@PTX to CAFs, the decrease of collagen fibre content, reduced the degree of tumour tissue fibrosis and opened up new ideas for anti-fibrosis therapy.
Ethics approval
All procedures involving laboratory animals are performed in accordance with the ethics committee guidelines at the Ocean University of China and Yantai University.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analysed in this study are included in this article.
Additional information
Funding
References
- Eso Y, Marusawa H. Novel approaches for molecular targeted therapy against hepatocellular carcinoma. Hepatol Res. 2018;48:597–607.
- Taitt HE. Global trends and prostate cancer: a review of incidence, detection, and mortality as influenced by race, ethnicity, and geographic location. Am J Mens Health. 2018;12:1807–1823.
- Fernandes C, Suares D, Yergeri MC. Tumor microenvironment targeted nanotherapy. Front Pharmacol. 2018;9:1230.
- Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. 2019;18:99–115.
- Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120.
- Wong KM, Horton KJ, Coveler AL, et al. Targeting the tumor stroma: the biology and clinical development of pegylated recombinant human hyaluronidase (PEGPH20. Current Oncology Reports. 2017;19:1–9.
- Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186–196.
- Santos AM, Jung J, Aziz N, et al. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. The Journal of Clinical Investigation. 2009;119:3613–3625.
- Bhuvaneswari R, Ng QF, Thong PS, et al. Nimotuzumab increases the anti-tumor effect of photodynamic therapy in an oral tumor model. Oncotarget. 2015;6:13487.
- Gollnick SO, Brackett CM. Enhancement of anti-tumor immunity by photodynamic therapy. Immunol Res. 2010;46(1-3):216–226.
- Fang J, Xiao L, Joo KI, et al. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. Int J Cancer. 2016;138:1013–1023.
- Ji T, Zhao Y, Ding Y, et al. Transformable peptide nanocarriers for expeditious drug release and effective cancer therapy via cancer‐associated fibroblast activation. Angewandte Chemie. 2016;128:1062–1067.
- Liu M, Song W, Huang L. Drug delivery systems targeting tumor-associated fibroblasts for cancer immunotherapy. Cancer Letters. 2019;448:31–39.
- Chen B, Dai W, Mei D, et al. Comprehensively priming the tumor microenvironment by cancer-associated fibroblast-targeted liposomes for combined therapy with cancer cell-targeted chemotherapeutic drug delivery system. J Control Release. 2016;241:68–80.
- Wang J, Li Q, Li X, et al. A novel FAPα-based Z-Gly-Pro epirubicin prodrug for improving tumor-targeting chemotherapy. Eur J Pharmacol. 2017;815:166–172.
- Rehman A, Jafari SM, Tong Q, et al. Drug nanodelivery systems based on natural polysaccharides against different diseases. Adv Colloid Interface Sci. 2020;284:102251.
- Naveen C, Shastri N R. Polysaccharide nanomicelles as drug carriers[M]//Polysaccharide Carriers for Drug Delivery. Woodhead Publishing, 2019: 339–363.
- Zheng Y, Xie Q, Wang H, et al. Recent advances in plant polysaccharide-mediated nano drug delivery systems. Int J Biol Macromol. 2020;165(Pt B):2668–2683.
- Zhang X, Zhao M, Cao N, et al. Construction of a tumor microenvironment pH-responsive cleavable PEGylated hyaluronic acid nano-drug delivery system for colorectal cancer treatment. Biomater Sci. 2020;8:1885–1896.
- Liu Y, Wu F, Ding Y, et al. Preparation and characterization of paclitaxel/chitosan nanosuspensions for drug delivery system and cytotoxicity evaluation in vitro. Advanced Fiber Materials. 2019;1:152–162.
- Clement J, Rae A. Therapeutic drug combinations and delivery systems comprising c-raf kinase antisense polynucleotides for treating ocular diseases and disorders: U.S. Patent Application 11/753,900[P]. 2007-11-29.
- Guo C, Hou X, Liu Y, et al. Novel chinese angelica polysaccharide biomimetic nanomedicine to curcumin delivery for hepatocellular carcinoma treatment and immunomodulatory effect. Phytomedicine. 2021;80:153356.
- Wang Y, Xiao J, Duan Y, et al. Lycium barbarum polysaccharide ameliorates sjögren’s syndrome in a murine model. Mol Nutr Food Res. 2021;65(11):2001118.
- Chen D, Lian S, Sun J, et al. Design of novel multifunctional targeting nano-carrier drug delivery system based on CD44 receptor and tumor microenvironment pH condition. Drug Deliv. 2016;23:798–803.
- Spadea A, Rios de la Rosa JM, Tirella A, et al. Evaluating the efficiency of hyaluronic acid for tumor targeting via CD44. Mol Pharmac. 2019;16:2481–2493.
- Bousoik E, Qadri M, Elsaid KA. CD44 receptor mediates urate crystal phagocytosis by macrophages and regulates inflammation in a murine peritoneal model of acute gout. Sci Rep. 2020;10:1–15.
- Thorne RF, Wang Y, Zhang Y, et al. Evaluating nuclear translocation of surface receptors: recommendations arising from analysis of CD44. Histochem Cell Biol. 2020;153:77–87.