
ABSTRACT
Of the more than 190 distinct species of Mycobacterium genus, many are economically and clinically important pathogens of humans or animals. Among those mycobacteria that infect humans, three species namely Mycobacterium tuberculosis (causative agent of tuberculosis), Mycobacterium leprae (causative agent of leprosy) and Mycobacterium abscessus (causative agent of chronic pulmonary infections) pose concern to global public health. Although antibiotics have been successfully developed to combat each of these, the emergence of drug-resistant strains is an increasing challenge for treatment and drug discovery. Here we describe the impact of the rapid expansion of genome sequencing and genome/pathway annotations that have greatly improved the progress of structure-guided drug discovery. We focus on the applications of comparative genomics, metabolomics, evolutionary bioinformatics and structural proteomics to identify potential drug targets. The opportunities and challenges for the design of drugs for M. tuberculosis, M. leprae and M. abscessus to combat resistance are discussed.
Mycobacteria: the global disease burden
Mycobacteria belong to the genus Mycobacterium, which is the only genus representing the Mycobacteriaceae family (order: Actinomycetales; class: Actinobacteria). The genus was proposed in 1896, to include two species namely tubercle bacillus (now known as Mycobacterium tuberculosis) and leprosy bacillus (Mycobacterium leprae) [Citation1]. Currently, there are >190 distinct species of Mycobacterium genus, some of which are economically and clinically important pathogens of humans or animals [Citation2].
Among the human mycobacterial infections, those caused by M. tuberculosis (tuberculosis), M. leprae (leprosy) and Mycobacterium abscessus (chronic pulmonary infections) pose a public health concern. Mycobacterial infections affect ∼11–14 million people each year globally and tuberculosis (TB) alone is responsible for ∼1.3 million deaths each year, of which 374,000 were people living with HIV/AIDS. In 2016, 10.4 million people living with HIV/AIDS, were diagnosed with TB [Citation3].
In case of M. leprae infections, the World Health Organization (WHO) reported ∼200,000 new cases of leprosy each year (http://www.who.int/wer/2017/wer9235/en/). In 2016, 214,783 new cases of leprosy were reported globally. India reported the highest number (135,485) and Brazil 25,218 cases. Lack of effective early diagnostic tool(s), vaccines and limited understanding of the patterns of transmission contribute to the ongoing incidence.
In addition to TB and leprosy, Non-Tuberculous Mycobacteria (NTM), such as M. abscessus complex, are emerging worldwide as the cause of chronic pulmonary infections [Citation4,Citation5]. Incidence rates of pulmonary infections due to NTM, vary greatly with geographical regions. The global incidence remained 1.0–1.8 in a population of 100,000, however, these rates are much higher in the US, Western Pacific and Canada [Citation5]. The prevalence of NTMs has increased from 1.3% [Citation6] to 32.7% in Colorado, in cystic fibrosis patients [Citation7].
The growing challenge of antimicrobial resistance
The past few decades have seen a “discovery void” pertaining to antibacterial drug development, where very few new molecules have been patented or approved for clinical use [Citation8]. This is particularly true for drug discovery against mycobacteria, where no new drugs that were specifically developed for this purpose reached the clinic after the early 1960s until recently [Citation9]. Importantly, the emergence of resistance towards first-line and second-line drugs poses additional challenges to the development of suitable drugs in each of M. tuberculosis, M. leprae and M. abscessus. The approval of drugs and causes of resistance have been systematically reviewed for M. tuberculosis [Citation10,Citation11], M. abscessus [Citation12] and M. leprae [Citation13]. Antibiotic resistance can arise due to physiological, intrinsic or acquired factors [Citation10,Citation14]. The intrinsic resistance is attributed to cell-wall permeability, drug efflux systems, drug targets with low affinity and enzymes that neutralize drugs in the cytoplasm. The acquired resistance is conferred by chromosomal mutations.
Although the TB mortality rate is falling at ∼3% per year globally, drug-resistant TB remains a continuing threat [Citation3]. The spread of drug-resistant strains of TB (mono-resistant, multidrug-resistant, extensively drug-resistant and totally drug-resistant) is alarmingly high and accounted for 490,000 cases of multidrug-resistant TB (MDR-TB) in 2016. Around 47% of the MDR-TB cases are reported in Southeast Asia [Citation15]. Increase in isoniazid-susceptible rifampin resistance was also noted in 2016, with 110,000 cases globally. The global burden of MDR-TB has recently increased by >20% annually [Citation16] and the treatment is successful in only 50% of the MDR-TB cases. With the ongoing transmission of the drug-resistant strains of M. tuberculosis in communities, it is increasingly important to research novel drug targets and identify potential leads that can be expanded to new drugs.
Drug resistance in leprosy is diagnosed by the mouse footpad method, which is time and labour-intensive. Alternatively, mutations can be detected in drug-resistance-determining regions of M. leprae drug targets such as dihydropteroate synthase (for the drug: dapsone), β subunit of RNA polymerase (rifampin) and subunit A of DNA gyrase (ofloxacin). As drug resistance has been suspected only in cases that self-report at the hospital as a result of reactivation or relapse in leprosy, the numbers are currently low; however, these may increase if a field-based surveillance system is implemented.
M. abscessus is naturally resistant to many first-line antimicrobials, including all the current TB drugs. The acquired resistance to aminoglycosides is known to be due to mutations in genes such as rrs [Citation12]. Likewise, mutations in 23S rRNA and genes such as erm(41) and rrl are known to cause macrolide resistance in M. abscessus [Citation17].
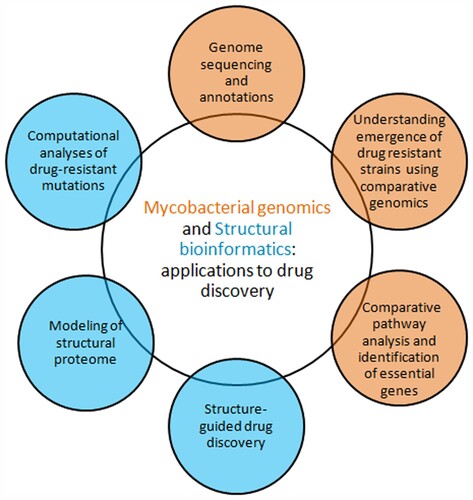
Thus, emergence of antibiotic-resistant strains and rapid spread due to globalization poses a serious challenge to the global health [Citation18]. Genomics has served as an important milestone in bacterial drug discovery [Citation19,Citation20]. It is now possible to understand causes of emergence of antibiotic-resistant strains and to identify potential drug targets through combinatorial approaches involving comparative genomics, metabolomics, phylogenomics, evolutionary and structural biology/bioinformatics (). Here we discuss the current status of drug discovery research in each area, focusing on the scope and applicability of computational approaches.
Figure 1. From mycobacterial genomes to drug discovery. Post-genomic application areas in mycobacterial drug discovery such as comparative genomics and structural biology/bioinformatics are shown.
Availability of genome sequences and annotation data: applications of comparative genomic approaches to drug discovery research
Comprehensive understanding of the organization of mycobacterial genomes began in 1998 with the elucidation of the complete genome sequence of M. tuberculosis [Citation21,Citation22]. The M. tuberculosis reference genome (H37Rv strain) is 4.41 Mbp in length and comprises 4081 protein genes, 13 pseudogenes, 45 tRNA genes, 30 ncRNA, 3 rRNA genes and 2 miscRNA genes (http://svitsrv8.epfl.ch/tuberculist/).
The genome of M. leprae was first sequenced in 2001 [Citation23]. This 3.2 Mbp genome has significant sequence similarity with that of M. tuberculosis; however, M. leprae reductively evolved to survive with 1614 protein genes, 1310 pseudogenes, 45 tRNA genes, 3 rRNA genes and 2 stable RNA genes (http://svitsrv8.epfl.ch/mycobrowser/leprosy.html).
The complete genome of M. abscessus strain ATCC 19977 was first sequenced in 2008 [Citation24]. The 5.06 Mbp genome consists of 4941 genes encoding 2886 proteins with functional assignments and 2055 hypothetical proteins (https://www.patricbrc.org/view/Genome/36809.5).
Rapid annotation of genomic data, leading to the development of general purpose as well as specialized resources, described in Supplementary Table 1, is providing important information pertinent to sequence, structure, function, metabolic pathway, taxonomy and drug resistance mutations.
Comparative genomics: understanding strain diversity and emergence of drug-resistant strains
Most pathogenic mycobacterium species including M. tuberculosis and M. leprae are slow growers taking >7 days to form visible colonies on solid media. Comparative pan-genomic analyses indicate that the evolution of rapid and slow growers is attributed to a series of gene gain and gene loss events leading to adaptation to different environments [Citation25]. Classical methods for genotyping of mycobacterial strains include IS6110DNA fingerprinting, spoligotyping and 24 locus-MIRU (mycobacterial-interspersed repetitive units)-VNTR (variable number of tandem repeats) typing [Citation26]. With the availability of genome sequencing data, complete genome-based phylogenies are being used for genotyping and classification of mycobacteria [Citation27,Citation28]. The classical M. tuberculosis complex is subdivided into seven distinct lineages with characteristic geographic distribution [Citation29]. Genomic analyses revealed three subspecies of M. abscessus, namely M. abscessus, M. massiliense and M. bolleti [Citation30]. Likewise, lineage diversity within distinct subtypes of M. leprae has been recently studied based on phylogenomic analyses of 154 genomes [Citation31].
Molecular phylogenetics and evolutionary dynamics methods allow study of epidemiology and links between genetic diversity and emergence of drug resistance in mycobacterial strains [Citation32]. Antibiotic-resistant genes are important markers for delineation of evolution and spread of drug resistance [Citation10,Citation14,Citation33]. Such computational studies have helped to answer specific questions such as: Whether a particular phylogenetic lineage is associated with drug resistance in an outbreak/epidemic? When and how the drug-resistant strains have emerged? What are the evolutionary factors that cause antimicrobial drug resistance in mycobacterium? Computational studies on mycobacterium addressing each of these questions are described below.
Recently, genomic studies focusing on population structure, origin and spread of MDR-TB have been undertaken across various parts of the world [Citation29,Citation34–36]. A study [Citation34] on strain diversity and phylogeography using genomes of 340 M. tuberculosis strains (isolated during 2008–2013) from KwaZulu-Natal, helped to elucidate the timing of acquisitions of drug resistance mutations that confer XDR-TB, indicating that isoniazid-resistance evolved earlier than rifampicin-resistance. Similarly, molecular epidemiology of MDR-TB in Ireland has been systematically examined using genomes of M. tuberculosis strains isolated during 2001–2014 from MDR-TB cases [Citation35]. Among seven lineages of MTB complex, Beijing lineage was observed to be associated with MDR [Citation35]. MDR-TB in Ireland was found to be introduced from other localities, as known for several European countries [Citation37].
In the case of M. abscessus, phylogenomic studies revealed the major role of recombination in causing lineage diversity [Citation30,Citation38,Citation39]. Population structure and recombination analyses provided significant evidence of gene flow and admixture among three lineages (M. abscessus, M. massiliense, and M. bolleti), and a correlation with pathogenicity and macrolide resistance in cystic fibrosis patients was found [Citation39]. Phylogenomic and genetic polymorphism analyses have also been carried out using M. abscessus isolates from US [Citation40]. A population genomic study [Citation41], based on the worldwide collection of clinical isolates of M. abscessus, has shown that the majority of M. abscessus infections are acquired through transmission (potentially via aerosols and fomites) of recently emerged circulating clones that have spread across the world. These clones are observed to be associated with increased virulence and worse clinical outcomes. This is a wake-up alarm! We are facing a pressing international infection challenge [Citation41].
In the case of M. leprae, a comprehensive study on phylogenomics and antibiotic resistance has been recently published [Citation31], which focuses on sequence and selection pressure analysis of wildtype as well as antibiotic-resistant genes. Several attempts have also been made to analyse the origin and spread of leprosy across various parts of the world [Citation42].
Thus, comparative genomics and phylogeographic studies are important in understanding the global spread and transmission dynamics of the mycobacterial infections. Strain diversity is shown to be one of the contributing factors to antibiotic resistance, linking transmission dynamics to medicine, across various geographic areas which are endemic for diseases such as TB [Citation43]. Furthermore, all these developments point to an increasing need for new and effective drugs to combat antibiotic resistance. Ideally, new drugs must have a novel mode of action to reduce events of cross-resistance, optimal dose-response, pharmacokinetic and toxicity profiles allowing safe and short duration of therapy either solely or in combination with other drugs. Comparative genomics of metabolic pathways provide a means to rapidly identify potential gene targets and thus could aid in drug discovery research (detailed in next section).
Comparative pathway analyses and identification of essential genes
M. tuberculosis, M. abscessus and M. leprae survive in distinct ecological niches with distinct evolving features that enable them to adapt to their specific environments. Malhotra et al. [Citation44] describe a 90% overlap of known and proposed drug targets against all mycobacterial species. The study identified the major pathways such as chorismate, purine/pyrimidine and amino acid biosynthetic pathways, essential for the survival of mycobacteria, as conserved in all these three species with minor differences in the carbon metabolism pathways.
M. tuberculosis is known for its peculiar ability to survive in the human macrophage despite varying stress conditions (redox, acidic, and nitrosamine) within the host. This has contributed to a very elaborate DNA repair and recombination system. The most up-to-date metabolic reconstruction for M. tuberculosis describes 1228 metabolic reactions, 1011 genes and 998 metabolites [Citation45], where 250 protein-coding genes present in the lipid biosynthetic pathways are responsible for the thick cell wall. Mutations in these pathways give rise to further changes in their metabolic pathways.
Draft metabolic reconstruction for M. abscessus exists in BioCyc [Citation46] and KEGG [Citation47] databases, with 110 pathways including biosynthesis, metabolism, biodegradation and information processing pathways. Evolutionary analyses showed that M. abscessus is more closely related to other NTMs such as M. avium complex and has a well-supported membrane transport system with a large number of efflux pumps, resulting in multidrug-resistant features [Citation48]. As an opportunistic bacterium, M. abscessus is able to survive outside its host and also contains metabolic pathways not associated with pathogenesis, which contribute to its larger genome size as compared to obligate pathogens.
In case of M. leprae, reductive evolution has led to the loss of common catabolic pathways such as lipolysis and impairment of the energy metabolism pathways [Citation23]. Moreover, the production of cognate and prosthetic groups from transport, biosynthetic and electron transfer pathways is also affected [Citation49]. With the loss of several functions, and the presence of a large number of conserved but unknown functions of pseudogenes, further studies to characterize the metabolic pathways in M. leprae are required.
Approaches to identify essential genes and prioritization of drug targets
The availability of genome sequences of mycobacteria, and annotations in sequence, structure and pathway databases facilitate systems-level analysis wherein comparative genomics and evolutionary bioinformatics approaches can be integrated to identify the minimal set of essential genes, leading to faster identification of putative drug targets. Essential genes that are necessary for the survival of the bacterium, and are critical components of metabolic and physico-chemical pathways, can be identified by gene knockouts, saturation transposon mutagenesis, RNA interference, etc. Essential genes have been characterized experimentally in the case of M. tuberculosis [Citation50–52]. However, limited studies have been carried out in M. abscessus and M. leprae. Specifically, M. leprae cannot be cultured in vitro and thus demands computational identification of essential genes.
Computational approaches for identification of essential genes, including machine learning, flux balance analyses and comparative genomics [Citation53], are faster and cheaper than experimental methods [Citation53]. Machine learning methods utilize unique genomic features of essential genes such as length of proteins, codon usage, GC content, sub-cellular localization, higher rate of evolutionary conservation, etc. Dedicated resources such as Database of Essential Genes [Citation54] and the database of Online Gene Essentiality [Citation55] have been developed, which serve as a platform for identification and prioritization of essential drug targets in various species including M. leprae [Citation56]. Advent of next-generation sequencing has enabled design of comparative genomics workflows to identify essential genes in case of M. tuberculosis [Citation57].
Upon identification of a set of essential genes, prioritization of drug targets can be achieved by further screening of essential genes based on computational predictions of ADMET (absorption, distribution, metabolism, excretion and toxicity) properties, sub-cellular localization, etc. In view of growing concern about drug resistance, attempts have been made to prioritize drug targets based on various analyses such as identification of uniqueness in metabolome and similarities to known druggable proteins, analysis of the protein-protein interactome, flux balance analysis of reactome and sequence-structural analysis of targetability of genes using integrative strategies [Citation58,Citation59]. As most critical targets in a pathogenic organism are expected to be evolutionarily conserved, this has been applied to filter essential genes in M. tuberculosis [Citation58]. Likewise, convergent positive selection analyses were used to identify genes that possibly cause drug resistance in M. tuberculosis [Citation59]. Evolutionary rate has been proposed as a useful parameter for ranking and prioritizing antibacterial drug targets [Citation60]. Thus, understanding evolution of drug targets is one of the important aspects of the drug discovery, especially to tackle the problem of drug resistance.
Structural biology and bioinformatics to combat mycobacterial infections
Early approaches to the discovery of new antibiotics relied almost entirely on whole-cell phenotypic screening of natural products, microbial extracts and fermentation broths. This approach has helped in the discovery of most antibiotics in use to date and recently led to the approval of two new drugs, bedaquiline and delamanid, for the treatment of drug-resistant strains of TB [Citation61]. Structure-guided drug discovery, pioneered in academia and some companies in the 1970s became central to the discovery of antihypertensives that targeted renin and AIDS antivirals that targeted HIV protease in the 1980s and 1990s [Citation62–64]. Opportunities for structure-guided drug discovery of mycobacterial targets became real only later when genome sequences became available. Understanding the structure and mechanism of the target allowed progress and optimization of hit-to-lead molecules, as well as a better understanding of resistance mechanisms, identifying causes of potential side effects and drug-drug interactions. This understanding led many research groups to perform high-throughput screening (HTS) and fragment-based screening campaigns of chemical libraries [Citation65] directly against carefully selected targets of interest. The applications of structure-guided drug discovery to mycobacteria have been reviewed earlier [Citation66–68].
Structural features can also be used to further refine target selection and validation including lack of structural homology to human host to avoid mechanism-based toxicity and ligandability leading to modulation of target activity. Where the target protein has not been structurally or functionally characterized, it’s structural model can be built based on homologous proteins using programs like MODELLER [Citation69]. Important information regarding target protein function and properties can be obtained from databases such as TubercuList (http://svitsrv8.epfl.ch/tuberculist/) or CHOPIN [Citation70]. Further, druggability of targets can be predicted by analysing properties and depths of various pockets/hotspots (capable of small-molecule binding interactions), using several databases and programs [Citation71].
Fragment-based drug discovery (FBDD): a promising alternative to HTS approach
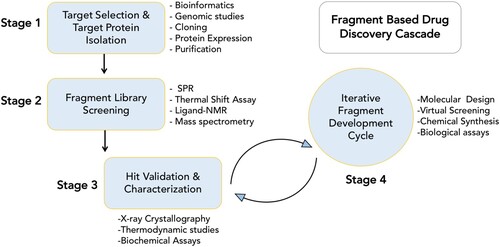
The principal advantage of the FBDD approach compared to HTS methods is that very small libraries (<103 fragments) of low molecular weight (<300 Da) compounds can be used to obtain good starting points for lead discovery [Citation65]. Initial fragment hits usually exhibit lower potency than the more complex molecules found in typical HTS compound libraries, but can be chemically optimized into lead candidates, thereby more effectively exploring the chemical space available for binding to the target protein [Citation68]. illustrates a typical fragment screening cascade employed in our lab. The details of these steps are summarized in Supplementary File 1.
Figure 2. FBDD cascade. Various techniques involved in each of the four stages are shown.
FBDD approaches have been successfully applied to design inhibitors targeting various key M. tuberculosis enzymes such as pantothenate synthetase [Citation72], transcriptional repressor [Citation73], cytochrome P450 [Citation74], thymidylate kinase [Citation75] and malate synthase [Citation76]. Some of the lead molecules developed from such fragment-based campaigns showed promising inhibitory response against M. tuberculosis, as shown in the example in Supplementary Figure 1.
Virtual screening, hotspot mapping and pharmacophore modelling to aid structure-guided drug discovery
In-silico screening and docking of compound libraries often help to reduce the time and cost involved in experimental testing of large sets of compounds to identify potential hits. The effectiveness of such virtual screening exercises can be improved by complementing the analysis with fragment hotspot-mapping programs [Citation71], which identify regions within the protein that provide relatively large contribution towards ligand binding in addition to information on interactions governing the predicted regions. Several studies [Citation77,Citation78] have used an energy-based pharmacophore modelling approach to complement virtual screening, followed by chemical optimization to identify inhibitors. We have collaborated with Maria Paola Costi at the University of Modena in a similar study involving a combination of virtual screening and molecular dynamics simulation methods to identify novel chemical scaffolds targeting M. tuberculosis thymidylate synthase X [Citation79].
Phenotypic screening along with advances in chemical genetics and bioinformatics has allowed target-guided compound identification and optimization [Citation80], by utilizing target mechanism-based whole-cell screening or by genetic manipulation of the target phenotype, or by finding targets directly from phenotypic hits [Citation81]. The latter approach can employ various techniques such as whole genome sequencing to identify resistant mutants [Citation82,Citation83], transcriptional profiling [Citation84], chemical biology and metabolomics [Citation85] or in-silico methods [Citation86]. These advances in the field discussed above, coupled with a comprehensive understanding of the chemical space of mycobacterial drugs, hold promise of accelerating the process of mycobacterial drug discovery.
From genomes to proteomes: automated modelling of proteomes through structural genomics and bioinformatics
Methods to identify structures that are potentially similar to the protein in question (templates) have been developed in our laboratory [Citation87] and elsewhere (reviewed in [Citation88]), as well as methods such as MODELLER [Citation69] and Rosetta [Citation89] to leverage this similarity to build theoretical, comparative models based on target-template alignments. Community-wide efforts, such as Critical Assessment Structure Prediction [Citation90] have led to substantial improvements in the accuracy of these approaches, both in terms of template identification and alignment, as well as model building and refinement. However, most of these methods focus on producing a single, most likely structure of each protein.
Our group has developed a structural genomics resource for M. tuberculosis (H37Rv strain) called CHOPIN [Citation70] which provides an ensemble of predicted models in different conformational states. These models are generated through a homology modelling pipeline. Development of such structural resources is important to identify potential therapeutic targets [Citation91].
Moving beyond proteomes: understanding the impact of drug-resistant mutations in drug targets
Mutations are likely to confer drug resistance by altering the energy landscape of the target protein, affecting the protein-protein interactions or affecting the drug/ligand binding with the target protein. Computational approaches to predict the effects of mutations on the structure and function of proteins can prove helpful in understanding the mechanism of drug resistance. Our lab has developed two well-established methods, SDM (Site Directed Mutator), based on a statistical approach using Environment Specific Substitution Tables [Citation92] and mCSM (mutation Cutoff Scanning Matrix), a machine learning approach [Citation93] to predict the structural and functional effects of mutations on the target proteins. mCSM is available in different flavours to predict the effects of mutations on protein stability (mCSM-stability), protein-protein interactions (mCSM-PPI) and protein-ligand interactions (mCSM-lig) [Citation94]. Additionally, in order to determine the impact of mutations on flexible protein conformations, tools like EnCOM [Citation95] and FoldX [Citation96] have been developed. Such methods have been used by our group and others to gain insights into the mechanism of mutations in various genetic and mycobacterial diseases including leprosy [Citation97–100]. Such analyses of the structural and functional mechanism of drug resistance causing mutations using computational approaches are very helpful in the rapid assessment of many mutations not easily achieved using experimental methods.
Challenges and future perspectives
This review has focused on the importance of advances in sequence and structure determination of genomes and their protein products in efforts to develop structure-guided drug discovery against three mycobacteria: M. tuberculosis, M. abscessus and M. leprae. Although M. tuberculosis is the most studied mycobacterium, 3D structures of only ∼16% of gene products have been determined experimentally. Sequence-structure homology recognition and comparative modelling have proved useful in providing clues about druggability. Furthermore, the more accurate models, based on structures of close homologues, can provide a basis for virtual screening and ligand design.
Metabolic reconstructions are providing insights into how M. tuberculosis has adapted within the host, and similar studies in M. abscessus and M. leprae are needed to provide better understanding of pathogenesis and design of new treatment regimes. The current focus in many laboratories is on providing user-friendly databases for the structural proteomes, and extending these not only to structures of homo- and hetero-oligomers, and complexes with natural and synthetic ligands, but also to incorporate data on metabolic pathways and epistasis, which are important in target selection.
The target-guided approach for antibiotic drug development has generally led to a low rate of translation of in-vitro inhibitory response to bactericidal/bacteriostatic activity. This challenge requires further study not only of physico-chemical properties of compounds affecting permeability of the thick waxy cell envelopes of mycobacteria, but also of drug efflux pumps and metabolic inactivation of compounds by bacterial/host cell enzymes that affect compound bioavailability. The emergence of drug resistance through mutations in the target is also a growing challenge. We have developed systematic mutagenesis studies using our software with a view to identify regions where drug design might be directed with a minimal chance of resistance. This is likely to be a key component in drug discovery in future, especially through structure-guided fragment-based approaches, where choices of fragment elaboration or cross-linking can be made in the light of such analyses of mutability.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Shinnick T, Good R. Mycobacterial taxonomy. Eur J Clin Microbiol Infect Dis. 1994;13:884–901.
- Forbes BA. Mycobacterial taxonomy. J Clin Microbiol. 2017;55:380.
- WHO. Global tuberculosis report 2017; 2017.
- Prevots DR, Loddenkemper R, Sotgiu G, et al. Nontuberculous mycobacterial pulmonary disease: an increasing burden with substantial costs. Eur Respir J. 2017;49:1700374.
- Stout JE, Koh WJ, Yew WW. Update on pulmonary disease due to non-tuberculous mycobacteria. Int J Infect Dis. 2016;45:123–134.
- Smith M, Efthimiou J, Hodson ME, et al. Mycobacterial isolations in young adults with cystic fibrosis. Thorax. 1984;39:369–375.
- Rodman DM, Polis JM, Heltshe SL, et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am J Respir Crit Care Med. 2005;171:621–626.
- Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109.
- D’Ambrosio L, Centis R, Sotgiu G, et al. New anti-tuberculosis drugs and regimens: 2015 update. ERJ Open Res. 2015;1:00010-2015.
- Gygli SM, Borrell S, Trauner A, et al. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev. 2017;41:354–373.
- Dookie N, Rambaran S, Padayatchi N, et al. Evolution of drug resistance in Mycobacterium tuberculosis: a review on the molecular determinants of resistance and implications for personalized care. J Antimicrob Chemother. 2018;73:1138–1151.
- Nessar R, Cambau E, Reyrat JM, et al. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother. 2012;67:810–818.
- Saunderson PR. Drug-resistant M leprae. Clin Dermatol. 2016;34:79–81.
- Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433.
- Variava E, Martinson N. Drug-resistant tuberculosis: the rise of the monos. Lancet Infect Dis. 2018;18:705–706.
- Lange C, Chesov D, Heyckendorf J, et al. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology. 2018;23:656–673.
- Liu W, Li B, Chu H, et al. Rapid detection of mutations in erm(41) and rrl associated with clarithromycin resistance in Mycobacterium abscessus complex by denaturing gradient gel electrophoresis. J Microbiol Methods. 2017;143:87–93.
- Bell G, MacLean C. The search for ‘evolution-proof’ antibiotics. Trends Microbiol. 2017;26:471–483.
- Pucci MJ. Use of genomics to select antibacterial targets. Biochem Pharmacol. 2006;71:1066–1072.
- Mills SD. When will the genomics investment pay off for antibacterial discovery? Biochem Pharmacol. 2006;71:1096–1102.
- Cole S, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537.
- Brosch R, Gordon SV, Billault A, et al. Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect Immun. 1998;66:2221–2229.
- Cole S, Eiglmeier K, Parkhill J, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007.
- Ripoll F, Pasek S, Schenowitz C, et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PloS One. 2009;4:e5660.
- Wee WY, Dutta A, Choo SW. Comparative genome analyses of mycobacteria give better insights into their evolution. PloS One. 2017;12:e0172831.
- Schürch AC, van Soolingen D. Dna fingerprinting of Mycobacterium tuberculosis: from phage typing to whole-genome sequencing. Infect Genet Evol. 2012;12:602–609.
- Roetzer A, Diel R, Kohl TA, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013;10:e1001387.
- Walker TM, Ip CL, Harrell RH, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–146.
- Niemann S, Supply P. Diversity and evolution of Mycobacterium tuberculosis: moving to whole-genome-based approaches. Cold Spring Harb Perspect Med. 2014;4:a021188.
- Sassi M, Drancourt M. Genome analysis reveals three genomospecies in Mycobacterium abscessus. BMC Genomics. 2014;15:359.
- Benjak A, Avanzi C, Singh P, et al. Phylogenomics and antimicrobial resistance of the leprosy bacillus Mycobacterium leprae. Nat Commun. 2018;9:352.
- Borrell S, Trauner A. Strain variation in the Mycobacterium tuberculosis complex: its role in biology, epidemiology and control. In: Gagneux S, editor. Advances in experimental medicine and biology, Vol. 1019, Chapter 14. Cham: Springer; 2017. p. 263–279.
- Aminov RI, Mackie RI. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol Lett. 2007;271:147–161.
- Cohen KA, Abeel T, Manson McGuire A, et al. Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med. 2015;12:e1001880.
- Roycroft E, O’Toole RF, Fitzgibbon MM, et al. Molecular epidemiology of multi-and extensively-drug-resistant Mycobacterium tuberculosis in Ireland, 2001–2014. J Infect. 2018;76:55–67.
- Merker M, Kohl TA, Roetzer A, et al. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PloS One. 2013;8:e82551.
- De Beer J, Kodmon C, van der Werf M, et al. Molecular surveillance of multi-and extensively drug-resistant tuberculosis transmission in the European Union from 2003 to 2011. Euro Surveill. 2014;19:pii: 20742.
- Tan JL, Ng KP, Ong CS, et al. Genomic comparisons reveal microevolutionary differences in Mycobacterium abscessus subspecies. Front Microbiol. 2017;8:2042.
- Sapriel G, Konjek J, Orgeur M, et al. Genome-wide mosaicism within Mycobacterium abscessus: evolutionary and epidemiological implications. BMC Genomics. 2016;17:118.
- Davidson RM, Hasan NA, Reynolds PR, et al. Genome sequencing of Mycobacterium abscessus isolates from patients in the United States and comparisons to globally diverse clinical strains. J Clin Microbiol. 2014;52:3573–82.
- Bryant JM, Grogono DM, Rodriguez-Rincon D, et al. Population-level genomics identifies the emergence and global spread of a human transmissible multidrug-resistant nontuberculous mycobacterium. Science. 2016;354:751–757.
- Monot M, Honoré N, Garnier T, et al. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat Genet. 2009;41:1282.
- Perdigão J, Clemente S, Ramos J, et al. Genetic diversity, transmission dynamics and drug resistance of Mycobacterium tuberculosis in Angola. Sci Rep. 2017;7:42814.
- Malhotra S, Vedithi SC, Blundell TL. Decoding the similarities and differences among Mycobacterial species. PLoS Negl Trop Dis. 2017;11:e0005883.
- Kavvas ES, Seif Y, Yurkovich JT, et al. Updated and standardized genome-scale reconstruction of Mycobacterium tuberculosis H37Rv, iEK1011, simulates flux states indicative of physiological conditions. BMC Syst Biol. 2018;12:25.
- Caspi R, Billington R, Ferrer L, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–D480.
- Kanehisa M. ‘In silico’ simulation of biological processes: Novartis foundation symposium 247.Wiley Online Library; 2002. p. 91–103.
- Shanmugham B, Pan A. Identification and characterization of potential therapeutic candidates in emerging human pathogen Mycobacterium abscessus: a novel hierarchical in silico approach. PloS One. 2013;8:e59126.
- Singh P, Cole ST. Mycobacterium leprae: genes, pseudogenes and genetic diversity. Future Microbiol. 2011;6:57–71.
- Griffin JE, Gawronski JD, DeJesus MA, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251.
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84.
- DeJesus MA, Gerrick ER, Xu W, et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio. 2017;8:e02133-16.
- Peng C, Lin Y, Luo H, et al. A comprehensive overview of online resources to identify and predict bacterial essential genes. Front Microbiol. 2017;8:2331.
- Zhang R, Ou HY, Zhang CT. Deg: a database of essential genes. Nucleic Acids Res. 2004;32:271D–D272.
- Chen W-H, Lu G, Chen X, et al. Ogee v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res. 2017;45(D1):D940–D944.
- Uddin R, Azam SS, Wadood A, et al. Computational identification of potential drug targets against Mycobacterium leprae. Med Chem Res. 2016;25:473–481.
- Ghosh S, Baloni P, Mukherjee S, et al. A multi-level multi-scale approach to study essential genes in Mycobacterium tuberculosis. BMC Syst Biol. 2013;7:132.
- Kaur D, Kutum R, Dash D, et al. Data intensive genome level analysis for identifying novel, non-toxic drug targets for multi drug resistant Mycobacterium tuberculosis. Sci Rep. 2017;7:46595.
- Farhat MR, Shapiro BJ, Kieser KJ, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet. 2013;45:1183.
- Gladki A, Kaczanowski S, Szczesny P, et al. The evolutionary rate of antibacterial drug targets. BMC Bioinformatics. 2013;14:36.
- Nguta JM, Appiah-Opong R, Nyarko AK, et al. Current perspectives in drug discovery against tuberculosis from natural products. Int J Mycobacteriol. 2015;4:165–183.
- Sibanda B, Pearl L, Hemmings A, et al. Acta crystallographica section A C42-C42. Copenhagen: Munksgaard.
- Blundell TL. Structure-based drug design. Nature. 1996;384:23–26.
- Blundell TL. Protein crystallography and drug discovery: recollections of knowledge exchange between academia and industry. IUCrJ. 2017;4:308–321.
- Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nat Rev Drug Discov. 2002;1:45–54.
- Mendes V, Blundell TL. Targeting tuberculosis using structure-guided fragment-based drug design. Drug Discov Today. 2017;22:546–554.
- Malhotra S, Thomas SE, Montano BO, et al. Structure-guided, target-based drug discovery–exploiting genome information from HIV to mycobacterial infections. Postepy Biochem. 2016;62:262–272.
- Thomas SE, Mendes V, Kim SY, et al. Structural biology and the design of new therapeutics: from HIV and cancer to mycobacterial infections: a paper dedicated to John Kendrew. J Mol Biol. 2017;429:2677–2693.
- Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815.
- Ochoa-Montaño B, Mohan N, Blundell TL. Chopin: a web resource for the structural and functional proteome of Mycobacterium tuberculosis. Database. 2015; 2015. pii:bav026.
- Radoux CJ, Olsson TS, Pitt WR, et al. Identifying interactions that determine fragment binding at protein hotspots. J Med Chem. 2016;59:4314–4325.
- Hung AW, Silvestre HL, Wen S, et al. Optimization of inhibitors of Mycobacterium tuberculosis pantothenate synthetase based on group efficiency analysis. Chem Med Chem. 2016;11:38–42.
- Nikiforov PO, Blaszczyk M, Surade S, et al. Fragment-sized EthR inhibitors exhibit exceptionally strong ethionamide boosting effect in whole-cell Mycobacterium tuberculosis assays. ACS Chem Biol. 2017;12:1390–1396.
- Kavanagh ME, Coyne AG, McLean KJ, et al. Fragment-based approaches to the development of Mycobacterium tuberculosis CYP121 inhibitors. J Med Chem. 2016;59:3272–3302.
- Naik M, Raichurkar A, Bandodkar BS, et al. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): discovery of 3-cyanopyridone and 1, 6-naphthyridin-2-one as potent inhibitors. J Med Chem. 2015;58:753–766.
- Huang H-L, Krieger IV, Parai MK, et al. Mycobacterium tuberculosis malate synthase structures with fragments reveal a portal for substrate/product exchange. J Biol Chem. 2016;M116:750877.
- Poyraz OM, Jeankumar VU, Saxena S, et al. Structure-guided design of novel thiazolidine inhibitors of O-acetyl serine sulfhydrylase from Mycobacterium tuberculosis. J Med Chem. 2013;56:6457–6466.
- Devi PB, Samala G, Sridevi JP, et al. Structure-guided design of thiazolidine derivatives as Mycobacterium tuberculosis pantothenate synthetase inhibitors. Chem Med Chem. 2014;9:2538–2547.
- Luciani R, Saxena P, Surade S, et al. Virtual screening and X-ray crystallography identify non-substrate analog inhibitors of flavin-dependent thymidylate synthase. J Med Chem. 2016;59:9269–9275.
- Zuniga ES, Early J, Parish T. The future for early-stage tuberculosis drug discovery. Future Microbiol. 2015;10:217–29.
- Abrahams GL, Kumar A, Savvi S, et al. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem Biol . 2012;19:844–854.
- Aggarwal A, Parai MK, Shetty N, et al. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell. 2017;170:249–259.e25.
- Singh V, Donini S, Pacitto A, et al. The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect Dis. 2017;3:5–17.
- Boshoff HI, Myers TG, Copp BR, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–40184.
- Prosser GA, de Carvalho LP. Metabolomics reveal d-alanine: d-alanine ligase as the target of d-cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett. 2013;4:1233–1237.
- Mugumbate G, Mendes V, Blaszczyk M, et al. Target identification of mycobacterium tuberculosis phenotypic hits using a concerted chemogenomic, biophysical, and structural approach. Front Pharmacol. 2017;8:681.
- Shi J, Blundell TL, Mizuguchi K. Fugue: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257.
- Dorn M, e Silva MB, Buriol LS, et al. Three-dimensional protein structure prediction: methods and computational strategies. Comput Biol Chem. 2014;53:251–276.
- Song Y, DiMaio F, Wang RY-R, et al. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742.
- Moult J, Fidelis K, Kryshtafovych A, et al. Critical assessment of methods of protein structure prediction (CASP)—round XII. Proteins: Struct, Funct, Bioinf. 2018;86:7–15.
- Hassan SS, Tiwari S, Guimarães LC, et al. Proteome scale comparative modeling for conserved drug and vaccine targets identification in Corynebacterium pseudotuberculosis. BMC Genomics. 2014;15(S3).
- Worth CL, Preissner R, Blundell TL. Sdm—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222.
- Pires DE, Ascher DB, Blundell TL. Mcsm: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–342.
- Pires DE, Blundell TL, Ascher DB. mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep. 2016;6:29575.
- Frappier V, Chartier M, Najmanovich RJ. Encom server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015;43:W395–W400.
- Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388.
- Vedithi SC, Malhotra S, Das M, et al. Structural implications of mutations conferring rifampin resistance in mycobacterium leprae. Sci Rep. 2018;8:5016.
- Pires DE, Chen J, Blundell TL, et al. In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci Rep. 2016;6:19848.
- Forman JR, Worth CL, Bickerton GRJ, et al. Structural bioinformatics mutation analysis reveals genotype–phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins: Struct, Funct, Bioinf. 2009;77:84–96.
- Pandurangan AP, Ascher DB, Thomas SE, et al. Genomes, structural biology and drug discovery: combating the impacts of mutations in genetic disease and antibiotic resistance. Biochem Soc Trans. 2017;45:303–311.