
ABSTRACT
Macrolides such as erythromycin are the empirical treatment of Bordetella pertussis infections. China has experienced an increase in erythromycin-resistant B. pertussis isolates since they were first reported in 2013. Here, we undertook a genomic study on Chinese B. pertussis isolates from 2012 to 2015 to elucidate the origins and phylogenetic relationships of erythromycin-resistant B. pertussis isolates in China. A total of 167 Chinese B. pertussis isolates were used for antibiotic sensitivity testing and multiple locus variable-number tandem repeat (VNTR) analysis (MLVA). All except four isolates were erythromycin-resistant and of the four erythromycin-sensitive isolates, three were non-ptxP1. MLVA types (MT), MT55, MT104 and MT195 were the predominant types. Fifty of those isolates were used for whole genome sequencing and phylogenetic analysis. Genome sequencing and phylogenetic analysis revealed three independent erythromycin-resistant lineages and all resistant isolates carried a mutation in the 23S rRNA gene. A novel fhaB3 allele was found uniquely in Chinese ptxP1 isolates and these Chinese ptxP1-ptxA1-fhaB3 had a 5-fold higher mutation rate than the global ptxP1-ptxA1 B. pertussis population. Our results suggest that the evolution of Chinese B. pertussis is likely to be driven by selection pressure from both vaccination and antibiotics. The emergence of the new non-vaccine fhaB3 allele in Chinese B. pertussis population may be a result of selection from vaccination, whereas the expansion of ptxP1-fhaB3 lineages was most likely to be the result of selection pressure from antibiotics. Further monitoring of B. pertussis in China is required to better understand the evolution of the pathogen.
Introduction
Pertussis (whooping cough) is caused by the Gram-negative bacterium Bordetella pertussis. Macrolides, especially erythromycin, are the empiric treatment for pertussis. Although the first erythromycin-resistant case was found in the United States in 1994 [Citation1], only a few resistant pertussis cases have been reported globally [Citation2,Citation3]. However, China has seen a rapid increase in erythromycin-resistant B. pertussis since the first case reported in 2013 [Citation4–6]. There are two known mechanisms for resistance to erythromycin: the acquisition of erythromycin-resistant methylase (erm) genes resulting in high-level resistance [Citation7], and mutations in the 23S rRNA gene leading to structural changes that prevent the binding of erythromycin [Citation2]. Previous studies of erythromycin-resistant B. pertussis isolates in China showed that all resistant isolates contained 23S rRNA gene mutations [Citation5,Citation8]. Moreover, these erythromycin-resistant isolates are also resistant to other macrolides [Citation4,Citation6].
Whole cell vaccine (WCV) against pertussis was introduced in China in early 1960s. Two component [pertussis toxin (Ptx) and filamentous haemagglutinin (Fha)] acellular pertussis vaccine (ACV) started to replace WCV in 2007 and completely replaced WCV in 2012 [Citation6]. The national immunization programme for pertussis in China recommends three primary doses of DTaP (diphtheria, tetanus and acellular pertussis) at 3, 4 and 5 months and a booster at 18–24 months of age [Citation9]. Despite over 99% vaccine coverage in children in China, B. pertussis infection is still very common among children, adolescent and adults [Citation10].
Although pertussis is a vaccine-preventable disease, the disease still imposes a heavy burden on global public health. Different types of ACV with 1–5 components were used by different countries [Citation9,Citation11,Citation12]. Ptx is the major virulence factor in B. pertussis and one of the main components in ACV. It has been suggested that ACV-induced selection pressure results in polymorphisms on the genes encoding ACV components [Citation13]. To date, 18 pertussis toxin promoter (ptxP) alleles have been identified but only two, ptxP1 and ptxP3, were commonly observed. Since early 2000, isolates carrying the ptxP3 allele became the predominant ptxP type in many countries, including the Netherlands, Finland and Australia [Citation13–15]. By contrast, the Chinese pertussis isolates predominantly carried the ptxP1 allele [Citation16,Citation17].
In this study, we aimed to elucidate the genomic evolution of erythromycin-resistant Chinese B. pertussis isolates from 2012 to 2015 and evaluate their molecular epidemiological characteristics.
Material and methods
Bacterial isolates and susceptibility to antimicrobials
A total of 167 isolates were collected according to Chinese clinical case diagnostic criteria for pertussis from 5 provinces in the Midwest of China between 2012 and 2015 (Supplementary Table1). Isolates were cultured on Bordet-Gengou agar (Oxoid) supplemented with 10% horse blood at 37°C for 3–5 days and further tested by E-test for resistance to erythromycin. Staphylococcus aureus ATCC25923 and B. pertussis ATCC9797 were used as controls.
Multiple locus variable-number tandem repeat (VNTR) analysis (MLVA)
MLVA was performed following the procedures described by Schouls et al. [Citation18]. Five loci (VNTR1, VNTR3, VNTR4, VNTR5 and VNTR6) were amplified by PCR and the MLVA type (MT) was assigned using the B. pertussis MLVA database at https://www.mlva.net/. The minimum spanning tree was generated using GrapeTree [Citation19].
Isolates selection and whole genome sequencing
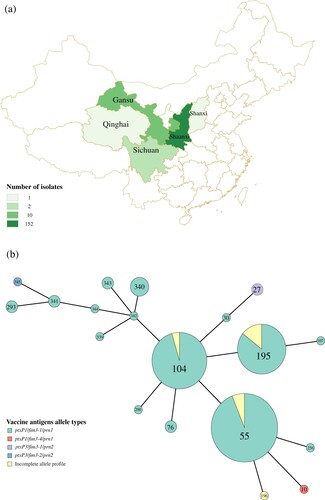
Real-time PCR targeting IS481 and ptxP were performed for all 167 suspected B. pertussis isolates. All isolates were found to be positive for both gene targets [Citation20]. Fifty of the 167 isolates were sequenced by Illumina paired end sequencing which included three ptxP3 isolates and 47 ptxP1 isolates representing different MTs (MT10, 27, 55, 76, 104, 195, 196, 256, 290, 293) from different years (2012, 2013, 2014 and 2015) and collected from 20 different cities in 5 provinces ((a)). Genomic DNA was extracted by the Wizard Genomic DNA Purification Kit (Promega). The isolates were sequenced using Illumina HiSeq and reads were assembled de novo using SPAdes version 3.7.0 [Citation21] (Supplementary Table2). Genomic DNA libraries were prepared using the KAPA HyperPlus Kit (Roche) and sequenced using HiSeq X Ten Reagent Kit v2.5 (Illumina). Raw reads were submitted to GenBank under the BioProject PRJNA489102.
Figure 1. (a) The geographical distribution of the 167 B. pertussis isolates. Regions were coloured by different shades proportional to the number of isolates from a given region as shown in the legend. (b) Minimum spanning tree of MLVA types of the 167 B. pertussis isolates collected between 2013-2015. Each circle represents an MLVA type (MT) with the number in the circle. The circle size is proportional to the number of isolates belonging to the particular MT. The fill colour of the circles indicates the different allelic profiles of vaccine antigen genes.
Analysis of vaccine antigen genes, 23S rRNA genes, and erythromycin ribosomal methylase (erm) genes
PCR was used for typing the vaccine antigen genes, ptxP, fim3 and prn [Citation22–24]. For isolates with sequenced genomes, in silico typing of the vaccine antigen genes, ptxP, ptxA, prn, fim2, fim3, and screening for 23S rRNA mutation and the presence of the erm genes [Citation25] were performed using BLAST+ tools (version 2.6.0). The reported A2047G position is based on an old Tohama I 23S rRNA sequence [Citation2] and is equivalent to A2037G in the current Tohama I genome (Accession No. NC_002929.2). To be consistent with published work, we use the A2047G convention as position reference throughout the text.
Single nucleotide polymorphism (SNP) detection and phylogenetic analysis
SNP detection used a combination of mapping by Burrow-Wheeler Alignment (BWA) tool version 0.7.12 [Citation26] and de novo assembly using SPAdes (version 3.7.0) [Citation21]. SAMtools version 0.1.19 [Citation27] was used for SNP calling. IGV version 2.3.94 was used to manually inspect read alignments to confirm SNP calls. Alignment of de novo assemblies was performed using progressiveMauve (version snapshot_2015_02-25). Phylogenetic trees were constructed using MEGA version 5.2.1, the maximum parsimony tree was applied based on the Tree-bisection-Reconnection (TBR) method and the bootstrap analysis was based on 1000 replicates (SNPs files shown in Supplementary Table3). Short insertions/deletions (indels) (less than 100 bp) were identified by SAMtools. Determination of IS481 and IS1002 insertion was performed using a custom script as previously described in Safarchi et al. [Citation28] and Octavia et al. [Citation29]. Estimation of the nucleotide substitution rate for each site was performed using Bayesian analysis by the BEAST package version 1.8.4 [Citation30]. The uncorrelated relaxed clock with constant population size model was found to be the most appropriate for Chinese B. pertussis lineages. The IEDB database (https://www.iedb.org/) [Citation31] was used for Fha epitope searching and the program PROVEAN (provean.jcvi.org/index.php) [Citation32] was used for predicting the effect of amino acid change.
Ethics
This study was approved by the Institutional Review Board of the Xi’an Center for Disease Control and Prevention, Xi’an, China.
Results
Genotypic analysis and antibiotic testing of B. pertussis isolates
A total of 167 isolates were collected from 2012 to 2015, from Midwest China ((a), Supplementary Table 1). Of these, 163 isolates had an erythromycin minimum inhibitory concentration (MIC) >256 IU/L suggesting erythromycin-resistance phenotype while the remaining four isolates were sensitive to erythromycin. Of these four erythromycin-sensitive isolates, one was a ptxP1 isolate and three were ptxP3 isolates.
Genotypic typing of vaccine antigen genes by PCR sequencing [Citation24] showed that 156 were ptxP1/fim3-1/prn1, ptxP1/fim3-4/prn1, while 3 were ptxP3/fim3-1/prn2, ptxP3/fim3-2/prn2. The remaining isolates contained incomplete allelic profiles ((b)). MLVA was performed on these isolates. Twelve MTs were observed of which seven were new (MT339-MT345). MT55, MT104 and MT195 were the predominant MTs, all of which except the incomplete allele profiles had ptxP1 allele. Of the three ptxP3 isolates, two belonged to MT27, a common ptxP3 MT, and one was a new type.
Whole genome sequencing of selected isolates
Fifty isolates including 47 ptxP1 isolates and three ptxP3 isolates were sequenced. Using Tohama I as the reference genome, the number of SNPs varied from 192 to 253 per isolate. Among the 47 ptxP1 Chinese isolates, 517 SNPs were detected, of which 33.46% (173/517) were non-coding (nc) SNPs (ncSNPs) located in intergenic (IG) regions, 21.86% (113/517) synonymous (s) SNPs (sSNPs) and 37.14% (192/517) non-synonymous (ns) SNPs (nsSNPs). Note that the three ptxP3 isolates were found to be distributed amongst global ptxP3 strains and were not analysed further (Supplementary Figure1).
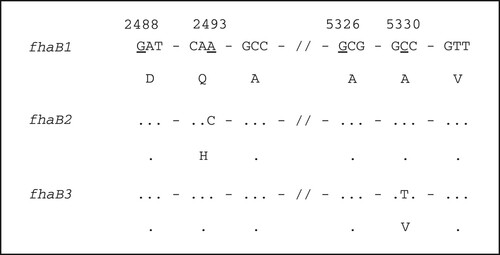
In silico analysis of the vaccine antigen genes was consistent with PCR sequencing results. Additionally, a mutation in position 5330 of the fhaB gene that resulted in the amino acid changing from alanine to valine was found in 46 isolates. Following the convention for fhaB allele naming by van Loo et al. [Citation33] which defined fhaB1 and fhaB2 alleles, we designated the novel allele with C5330 T mutation as fhaB3 (GenBank Accession No. MH824675) (). The mutated region was in one of the Fha epitopes according to the database IEDB (https://www.iedb.org/), which is related to the MHC class I. PROVEAN (http://provean.jcvi.org/index.php) predicting showed that the substitution does not affect protein structure. However, it may still affect immune recognition.
Figure 2. Sequence difference of different fhaB alleles. Numbers at the top refer to the position of the underline nucleotide, which is relative to the start of fhaB gene. Dots indicate identity. Nucleotide sequence is shown in codons with amino acid in single letter shown below.
All genomes were searched for the presence of erm genes, but none were found [Citation25]. Instead, the 46 erythromycin-resistant isolates contained a A2047G mutation in the domain V region of the 23S rRNA genes. B. pertussis has 3 copies of the rrn operon. No heterogenous bases were detected at 2047 site of the 23S rRNA gene in any of the 46 erythromycin-resistant isolates, suggesting that all three rrn operons carried the A2047G mutation.
Phylogenetic analysis of erythromycin-resistant Chinese B. pertussis isolates
A total of 71 Chinese B. pertussis isolates were used for phylogenetic analysis, including 47 ptxP1 isolates from this study and 24 ptxP1 isolates from a previous Chinese study [Citation34] (). Tohama I was used as the outgroup. The 46 erythromycin-resistant isolates were grouped into three lineages: Lineage I (18 isolates), Lineage II (12 isolates) and Lineage III (16 isolates), which were supported by five [one sSNP, one nsSNP and three ncSNPs], 12 (three sSNPs, five nsSNPs and four ncSNPs) and nine SNPs (three sSNPs, five nsSNPs and one ncSNP) (Supplementary Table4), respectively. Each of the three lineages were supported by a bootstrap value of 100% with 1000 replicates.
Figure 3. Phylogenomic relationship of 71 Chinese B. pertussis isolates. A maximum parsimony tree was generated based on 928 SNPs to illustrate the genetic relationship of erythromycin-resistant isolates. Tohama I was used as an outgroup. The three erythromycin-resistant lineages I, II, and III were marked by red, green and blue respectively. Bootstrap values of ≥50% were marked on each branch. The unit bar represents 10 SNPs. Isolate details (year, phenotype of and genotype of erythromycin-sensitivity and fhaB allele type) are shown as colour codes per the legends. MLVA types are shown in the brackets following the isolate's name.
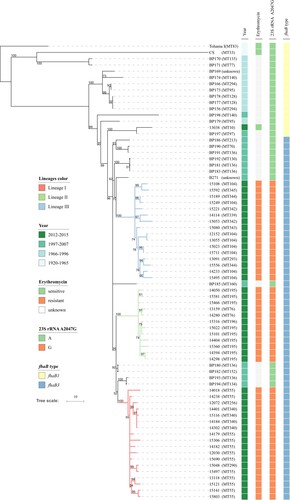
Lineage I was grouped together with 2 erythromycin-sensitive isolates BP193 and BP194, supported by one nsSNP. Lineages I and II were closely related, supported by two SNPs (one ncSNP and one sSNP), and grouped together with two erythromycin-sensitive isolates, BP180 and BP182. They were clearly separated from Lineage III, supported by a bootstrap value of 83%. Lineage III shared one sSNP with erythromycin-sensitive isolates BP181, BP183 and BP192 as well as an erythromycin-sensitive isolate from Taiwan (B271), supported by a bootstrap value of 62%. Bootstrap value for the erythromycin-resistant branches with interspersed erythromycin-sensitive isolates was 96%. These high bootstrap values support the inference that these erythromycin-resistance lineages were derived from erythromycin-sensitive isolates and erythromycin-resistance arose independently three times.
Relationships of erythromycin-resistant Chinese isolates with global ptxP1 isolates
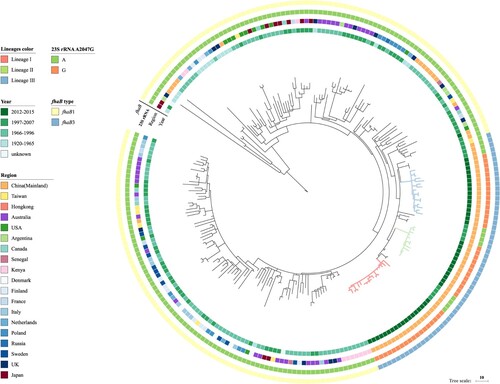
We expanded our genome analysis to include 162 global ptxP1 isolates from Bart et al. study [Citation15] (). The Chinese erythromycin-resistant isolates and 12 other Chinese erythromycin-sensitive isolates from 1997 to 2007 were grouped together exclusively without isolates from other regions, showing that the erythromycin-resistant lineages arose out of the erythromycin-sensitive isolates in China. There were three SNPs supporting this group (located in genes BP1879, BP3362 and BP3466). We named this group as the fhaB3 lineage as all isolates within the group had the fhaB3 allele (). No ptxP1 isolates, other than the 46 Chinese isolates from this study, had the A2047G mutation in the 23S rRNA gene suggesting that they were all erythromycin-sensitive. BEAST analysis showed the three clusters arose at different times: Lineage III arose around 2003 while Lineage I and II arose around 2008 and 2007, respectively.
Figure 4. Genomic relationship of global ptxP1 strains. The tree was made using 235 B. pertussis genomes including the 47 Chinese ptxP1 strains sequenced in this study and was based on 2744 SNPs by the maximum parsimony method. Three erythromycin-resistant lineages I, II, and III were marked by red, green and blue respectively. Tohama I was used as the outgroup. The unit bar represents 10 SNPs. Isolate details (year, region, genotype of erythromycin-sensitvity and fhaB allele type) are shown as colour codes as per the legends.
Insertion sequence (IS) elements
B. pertussis genome has more than 200 copies of insertion sequence (IS) elements [Citation35]. In the B. pertussis reference genome, Tohama I, it contains 238 and six copies of IS481, IS1002 respectively [Citation36]. In the 50 genomes from this study, the average copy number of IS481 and IS1002 was 197 and five, respectively. There was no unique IS insertion common to all erythromycin-resistant isolates. IS elements inserted in the prn gene have been shown to result in Prn negative isolates [Citation12]. One isolate (15221) was found to have an IS481 inserted at position 1598 in its prn gene, and thus was likely to be a Prn negative isolate.
Mutation rate
A previous study suggested different lineages may have different mutation rates. We calculated the mutation rate of the fhaB3 lineage and the mutation rate of ptxP1-ptxA1 strains from the global isolates excluding the fhaB3 isolates for comparison. Note that nearly all isolates were ptxA1 after the introduction of WCV in the 1950s and the genetic diversity of the B. pertussis population was reduced with the replacement of previously prevalent ptxA2 strains by ptxA1 strains [Citation14]. The mutation rate of the fhaB3 lineage was 7.22 × 10–3 (95%CI 4.78 × 10–3, 9.87 × 10–3), which was 1.21 × 10–6 substitution/site/year using a core genome size of 3,485,486 bp [Citation36], while the mutation rate of the global non-fhaB3 isolates was 5.39 × 10–4 (95%CI 4.48 × 10–4, 6.34 × 10−4) which translates to 2.11 × 10–7 substitution/site/year. Our estimate is similar to the earlier estimates of the Chinese B. pertussis population by Xu et al. [Citation34] of 3.06 × 10–6 substitution/site/year, confirming that the Chinese ptxP1 lineage evolves faster than the global ptxP1 population. Note that the mutation rate of 2.24 × 10–7 substitution/site/year by Bart et al. [Citation15] included ptxP3 strains and is an overall rate. We examined mutator genes for any mutations [Citation37] that may have contributed to the increased mutation rate. No SNPs were located in any of the mutator genes examined.
Discussion
This study has shown that ptxP1 B. pertussis strains continue to be more prevalent in China as 98.11% (156/159) of the isolates carried ptxP1 allele, which is different from the global trend where ptxP3 strains were more prevalent [Citation15,Citation16]. The vast majority of the isolates (97.6% [163/167]) were identified as erythromycin-resistant, showing rapid expansion of erythromycin-resistant strains in China.
Erythromycin-resistant Lineages I, II and III were represented predominantly by MT55, MT195 and MT104, respectively (Figure1 (b)). The three major MTs types were different from a previous study, which showed that the MLVA types of Chinese isolates have changed continuously in different periods. However, it is also possible that different B. pertussis populations circulate in different regions of China as most isolates in Xu et al. study were from Beijing in the North of China while isolates from this study were from Midwestern regions. Xu et al. also showed that the MLVA types of Chinese isolates changed dynamically in different periods [Citation16].
Non-expression of Prn has been reported in many countries where ACV containing Prn was used and high prevalence of Prn deficient strains have been reported in Australia and some other countries [Citation12,Citation38–40]. Interestingly, in this study, one isolate (15221) was found to have IS481 inserted in its prn gene. The insertion was at a location known to cause Prn inactivation and thus the isolate is most likely not expressing Prn. This Prn inactivation is unlikely to be due to selection pressure from the Chinese ACV as it contains no Prn. However, the emergence of the first Prn-negative isolate, which occurred in an erythromycin-resistant ptxP1 background, raises concerns of such isolates spreading to countries where ACVs containing Prn are used since higher fitness was shown for Prn negative strains in a mouse model [Citation41].
Phylogenetic analysis showed that erythromycin-resistant isolates in this study evolved as three independent lineages from erythromycin-sensitive strains. The three lineages were well supported by multiple SNPs. Each lineage was grouped with erythromycin-sensitive isolates, supported by one to two SNPs. Although it is possible that a SNP may arise independently, there have been no parallel mutations observed in B. pertussis except in vaccine antigen genes [Citation38]. The SNPs supporting the grouping of sensitive isolates with Lineage I (five) and Lineage II (12) were located in ncSNPs or sSNPs and thus less likely to be under positive selection pressure. Further, Lineages I and II, together with closely related erythromycin-sensitive isolates were well separated from Lineage III, providing a firm support of independent origin of these lineages.
All Chinese fhaB3 formed a separate lineage that contained only Chinese isolates, suggesting that this mutation arose once and expanded in China. The fhaB3 mutation arose in early 1990s from BEAST estimates, before ACV was introduced in China but ACV may have selected for the new allele. As the Chinese vaccine strain CS contains fhaB1 allele, the emergence and expansion of fhaB3 allele may be a result of selection pressure from vaccination. Additionally, the expansion of fhaB3 isolates may also be due to the selection pressure from antibiotics as the majority of the fhaB3 isolates from this study were resistant to erythromycin. Further studies are required to determine whether this mutation confers any advantage as Fha is one of the two components (Ptx and Fha) of the Chinese ACV. It is important to note that the Chinese vaccine strain CS contained ptxA2-fhaB1 alleles while the Chinese ptxP1 B. pertussis population carried ptxA1-fhaB3 alleles, causing mismatch in both vaccine antigen alleles.
The Chinese ptxP1 strains formed a single lineage which also uniquely carried the fhaB3 allele. The mutation rate of the Chinese ptxP1-ptxA1-fhaB3 lineage was five times higher than the rest of the global ptxP1-ptxA1 isolates, consistent with an earlier estimate. The genetic basis of increased mutation rate is unknown as there were no SNPs in any of the mutator genes that may increase mutation rate [Citation37]. The increased mutation rate may have played a role in the adaptation of Chinese B. pertussis against antibiotic-driven selection pressure and/or WCV selection pressure.
The rise of erythromycin/azithromycin resistance in B. pertussis in China could be from direct or indirect selection pressure from the use of macrolides. Sixteen isolates included in this study have previously been shown to be resistant to azithromycin by Li et al. [Citation42], suggesting that the erythromycin-resistant isolates in this study were also resistant to azithromycin. Macrolides are the primary therapeutic choice of both pertussis and Mycoplasma pneumoniae associated pneumonia. Macrolide-resistant M. pneumoniae in China has been previously reported with prevalence of resistance from 69% to 95% [Citation43,Citation44]. It is likely that the frequent use of erythromycin has contributed to the emergence and spread of both erythromycin-resistant M. pneumoniae and B. pertussis. There are few reports of erythromycin-resistant B. pertussis [Citation3,Citation5,Citation45,Citation46]. Resistance is low or none in European countries [Citation47], Australia [Citation48] and the US [Citation49] where the use of antibiotics is controlled and guided by microbiological diagnosis. It will be interesting to see how the B. pertussis population evolves in countries where there is a similar high usage of antibiotics to China. However, many developing countries still use the whole cell vaccine, where the selection pressures may be different from what we have seen in China.
It is intriguing that all erythromycin-resistant isolates had a high MIC of >256 μg/ml with no intermediate level of resistance observed. Lower resistance has been reported previously in two US isolates with one each displaying MIC values of 32 and 64 μg/ml [Citation3,Citation50]. The resistance in the Chinese isolates has been attributed to the A2047G mutation in the 23S rRNA gene while the mechanism of resistance in the US isolates is unknown. We found no other erythromycin resistance genes present in the genomes of the Chinese isolates. The presence of the resistance conferring A2047G mutation in all 3 copies of the 23S rRNA may have conferred the high level of erythromycin resistance. A likely explanation of how all 3 copies of the 23S rRNA gene shared the same mutation is homologous recombination between the rRNA operons. The A2047G mutation may have initially appeared in one copy of the 23S rRNA due to antibiotic selection and then recombined into the other 2 copies [Citation51].
The Chinese B. pertussis population predominantly carried the ptxP1 allele. It has been observed previously that there is little geographical separation of B. pertussis population [Citation15,Citation29]. Overall, ptxP3 B. pertussis circulates in countries where ACV has been in use while WCV countries have been dominated by ptxP1 strains [Citation52]. The replacement of WCV in China in 2012 occurred a decade later than developed countries such as the US and Australia [Citation6]. Therefore, there may not have been enough time for ptxP3 strains to replace ptxP1 strains in China. However, ptxP3 strains were predominant before ACV was introduced in some countries such as the US [Citation13]. Additionally, ptxP3 strains are fitter than ptxP1 strains regardless of ACV immunization in a mixed infection mouse model [Citation53] and thus are expected to outcompete ptxP1 strains in its natural hosts. The predominance of ptxP1 strains in China may be a result of selection from antibiotics as the erythromycin-resistant strains clearly have had an advantage over erythromycin-sensitive ptxP3 strains in a host population where antibiotics are known to be overused. Therefore, antibiotic-driven selection pressure may play a greater role in the evolution of B. pertussis isolates than vaccine in China and help maintain a separate B. pertussis population.
In addition to the much later introduction of ACV, China also uses an ACV with different ACV components and a different vaccine schedule. The Chinese national immunization programme recommends three primary doses of DTaP at 3, 4 and 5 months and a booster at 18–24 months [Citation9]. No pertussis booster doses are administered for primary school age children as the booster given to 6-year-old children only includes diphtheria and tetanus toxoids without the acellular pertussis component [Citation9]. Given the differences in vaccine and antibiotic selection pressures imposed on the Chinese B. pertussis population, we expect a different evolutionary trajectory of B. pertussis in China.
Conclusion
In conclusion, the evolution of Chinese B. pertussis is likely to be driven by selection pressure from both vaccination and antibiotics, which may have maintained the unique B. pertussis population in China. The potential spread of the erythromycin-resistant Chinese ptxP1 strains to other countries is also of global public health concern, especially if they gain a competitive advantage over ptxP3 strains currently circulating more globally. Further monitoring of B. pertussis in China is required to better understand the evolution of the pathogen.
Supplemental Material
Download Zip (548.4 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Choo V. Erythromycin-resistant Bordetella pertussis. The Lancet. 1994;344:1564. doi: 10.1016/S0140-6736(94)90365-4
- Bartkus JM, Juni BA, Ehresmann K, et al. Identification of a mutation associated with erythromycin resistance in Bordetella pertussis: implications for Surveillance of Antimicrobial resistance. J Clin Microbiol. 2003;41:1167–1172. doi: 10.1128/jcm.41.3.1167-1172.2003
- Korgenski EK, Daly JA. Surveillance and detection of erythromycin resistance in Bordetella pertussis isolates recovered from a pediatric population in the Intermountain West region of the United States. J Clin Microbiol. 1997;35:2989–2991.
- Wang Z, Li Y, Hou T, et al. Appearance of macrolide-resistant Bordetella pertussis strains in China. Antimicrob Agents Chemother. 2013;57:5193–5194. doi: 10.1128/AAC.01081-13
- Wang Z, Cui Z, Li Y, et al. High prevalence of erythromycin-resistant Bordetella pertussis in Xi’an, China. Clin Microbiol Infect. 2014;20:O825–O830. doi: 10.1111/1469-0691.12671
- Liu X, Wang Z, Zhang J, et al. Pertussis Outbreak in a primary school in China: infection and transmission of the macrolide-resistant Bordetella pertussis. Pediatr Infect Dis J. 2018;37:e145–e148.
- Pechère J-C. Macrolide resistance mechanisms in Gram-positive cocci. Int J Antimicrob Agents. 2001;18:25–28. doi: 10.1016/S0924-8579(01)00407-1
- Wang Z, Han R, Liu Y, et al. Direct detection of erythromycin-resistant Bordetella pertussis in clinical Specimens by PCR. J Clin Microbiol. 2015;53:3418–3422. doi: 10.1128/JCM.01499-15
- Zhang L, Xu Y, Zhao J, et al. Effect of vaccination on Bordetella pertussis strains, China. Emerg Infect Dis. 2010;16:1695–1701. doi: 10.3201/eid1611.100401
- Zhang Q, Zheng H, Liu M, et al. The seroepidemiology of immunoglobulin G antibodies against pertussis toxin in China: a cross sectional study. BMC Infect Dis. 2012;12:138. doi: 10.1186/1471-2334-12-138
- Thierry-Carstensen B, Dalby T, Stevner MA, et al. Experience with monocomponent acellular pertussis combination vaccines for infants, children, adolescents and adults – a review of safety, immunogenicity, efficacy and effectiveness studies and 15 years of field experience. Vaccine. 2013;31:5178–5191. doi: 10.1016/j.vaccine.2013.08.034
- Lam C, Octavia S, Ricafort L, et al. Rapid increase in pertactin-deficient Bordetella pertussis isolates, Australia. Emerg Infect Dis. 2014;20:626–633.
- Mooi FR, van Loo IH, van Gent M, et al. Bordetella pertussis strains with increased toxin production associated with pertussis resurgence. Emerg Infect Dis. 2009;15:1206–1213. DOI:10.3201/eid1508.081511.
- Octavia S, Maharjan RP, Sintchenko V, et al. Insight into evolution of Bordetella pertussis from comparative genomic analysis: evidence of vaccine-driven selection. Mol Biol Evol. 2011;28:707–715. doi: 10.1093/molbev/msq245
- Bart MJ, Harris SR, Advani A, et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio. 2014;5:e01074.
- Xu Y, Zhang L, Tan Y, et al. Genetic diversity and population dynamics of Bordetella pertussis in China between 1950–2007. Vaccine. 2015;33:6327–6331. doi: 10.1016/j.vaccine.2015.09.040
- Wang Z, He Q. Bordetella pertussis isolates circulating in China where whole cell vaccines have been used for 50 years. Clin Infect Dis. 2015;61:1028–1029. doi: 10.1093/cid/civ457
- Schouls LM, Van Der Heide HG, Vauterin L, et al. Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J Bacteriol. 2004;186:5496–5505. doi: 10.1128/JB.186.16.5496-5505.2004
- Zhou Z, Alikhan N-F, Sergeant MJ, et al. Grapetree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res 2018;28:1395–1404. doi: 10.1101/gr.232397.117
- Dragsted DM, Dohn B, Madsen J, et al. Comparison of culture and PCR for detection of Bordetella pertussis and Bordetella parapertussis under routine laboratory conditions. J Med Microbiol. 2004;53:749–754. doi: 10.1099/jmm.0.45585-0
- Nurk S, Bankevich A, Antipov D, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20:714–737. doi: 10.1089/cmb.2013.0084
- Van Gent M, Heuvelman C, Van der Heide H, et al. Analysis of Bordetella pertussis clinical isolates circulating in European countries during the period 1998–2012. Eur J Clin Microbiol Infect Dis. 2015;34:821–830. doi: 10.1007/s10096-014-2297-2
- Mooi F, Hallander H, Von König CW, et al. Epidemiological typing of Bordetella pertussis isolates: recommendations for a standard methodology. Eur J Clin Microbiol Infect Dis. 2000;19:174–181. doi: 10.1007/s100960050455
- Du Q, Wang X, Liu Y, et al. Direct molecular typing of Bordetella pertussis from nasopharyngeal specimens in China in 2012–2013. Eur J Clin Microbiol Infect Dis. 2016;35:1211–1214. doi: 10.1007/s10096-016-2655-3
- Choi J, Rieke EL, Moorman TB, et al. Practical implications of erythromycin resistance gene diversity on surveillance and monitoring of resistance. FEMS Microbiol Ecol. 2018;94, fiy006.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–wheeler transform. bioinformatics 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352
- Safarchi A, Octavia S, Wu SZ, et al. Genomic dissection of Australian Bordetella pertussis isolates from the 2008-2012 epidemic. J Infect. 2016;72:468–477. doi: 10.1016/j.jinf.2016.01.005
- Octavia S, Wu SZ, Kaur S, et al. Whole-genome sequencing and comparative genomic analysis of Bordetella pertussis isolates from the 2007–2008 epidemic in Israel. Journal of Infection 2017;74:204–207. doi: 10.1016/j.jinf.2016.11.012
- Suchard MA, Lemey P, Baele G, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4, vey016.
- Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2014;43:D405–D412.
- Choi Y, Chan P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195
- van Loo IHM, Heuvelman KJ, King AJ, et al. Multilocus sequence typing of Bordetella pertussis based on Surface protein genes. J Clin Microbiol 2002;40:1994–2001. doi: 10.1128/jcm.40.6.1994-2001.2002
- Xu Y, Liu B, Gröndahl-Yli-Hannuksila K, et al. Whole-genome sequencing reveals the effect of vaccination on the evolution of Bordetella pertussis. Sci Rep. 2015;5:12888. doi: 10.1038/srep12888
- Brinig MM, Cummings CA, Sanden GN, et al. Significant gene order and expression differences in Bordetella pertussis despite limited gene content variation. J Bacteriol. 2006;188:2375–2382. doi: 10.1128/JB.188.7.2375-2382.2006
- Parkhill J, Sebaihia M, Preston A, et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35:32. doi: 10.1038/ng1227
- Horst J-P, Wu T-H, Marinus MG. Escherichia coli mutator genes. Trends Microbiol 1999;7:29–36. doi: 10.1016/S0966-842X(98)01424-3
- Weigand MR, Peng Y, Loparev V, et al. The History of Bordetella pertussis genome evolution includes structural Rearrangement. J Bacteriol. 2017;199. doi: 10.1128/JB.00806-16
- Otsuka N, Han HJ, Toyoizumi-Ajisaka H, et al. Prevalence and genetic characterization of pertactin-deficient Bordetella pertussis in Japan. PLoS One. 2012;7:e31985. doi: 10.1371/journal.pone.0031985
- Barkoff AM, Mertsola J, Guillot S, et al. Appearance of Bordetella pertussis strains not expressing the vaccine antigen pertactin in Finland. Clin Vaccine Immunol. 2012;19:1703–1704. doi: 10.1128/CVI.00367-12
- Safarchi A, Octavia S, Luu LD, et al. Pertactin negative Bordetella pertussis demonstrates higher fitness under vaccine selection pressure in a mixed infection model. Vaccine 2015;33:6277–6281. doi: 10.1016/j.vaccine.2015.09.064
- Li Y, Liu X, Zhang B, et al. Where macrolide resistance is prevalent. Apmis. 2015;123:361–363. doi: 10.1111/apm.12357
- Cao B, Qu JX, Yin YD, et al. Overview of antimicrobial options for M ycoplasma pneumoniae pneumonia: focus on macrolide resistance. Clin Respir J. 2017;11:419–429. doi: 10.1111/crj.12379
- Zhao F, Lv M, Tao X, et al. Antibiotic sensitivity of 40 Mycoplasma pneumoniae isolates and molecular analysis of macrolide-resistant isolates from Beijing, China. Antimicrob Agents Chemother. 2012;56:1108–1109. doi: 10.1128/AAC.05627-11
- Shahcheraghi F, Lotfi MN, Nikbin VS, et al. The first macrolide-resistant Bordetella pertussis strains isolated from Iranian patients. Jundishapur J Microbiol. 2014;7(6):e10880.
- Yao S-M, Liaw G-J, Chen Y-Y, et al. Antimicrobial susceptibility testing of Bordetella pertussis in Taiwan prompted by a case of pertussis in a paediatric patient. J Med Microbiol. 2008;57:1577–1580. doi: 10.1099/jmm.0.2008/002857-0
- Lönnqvist E, Barkoff A-M, Mertsola J, et al. Antimicrobial susceptibility testing of Finnish Bordetella pertussis isolates collected during 2006–2017. J Glob Antimicrob Resist. 2018;14:12–16.
- Sintchenko V, Brown M, Gilbert GL. Is Bordetella pertussis susceptibility to erythromycin changing? MIC trends among Australian isolates 1971–2006. J Antimicrob Chemother. 2007;60:1178–1179. doi: 10.1093/jac/dkm343
- Souder E, Vodzak J, Evangelista AT, et al. Antimicrobial Susceptibility and molecular detection of pertactin-producing and pertactin-deficient Bordetella pertussis. Pediatr Infect Dis J. 2017;36:119–121. doi: 10.1097/INF.0000000000001366
- Centers for Disease Control and Prevention. Erythromycin-resistant Bordetella pertussis – Yuma County, Arizona, May–October 1994. MMWR Morb Mortal Wkly Rep. 1994;43:807.
- Ross JI, Eady EA, Cove JH, et al. Clinical resistance to erythromycin and clindamycin in cutaneous propionibacteria isolated from acne patients is associated with mutations in 23S rRNA. Antimicrob Agents Chemother. 1997;41:1162–1165. doi: 10.1128/AAC.41.5.1162
- Mosiej E, Zawadka M, Krysztopa-Grzybowska K, et al. Sequence variation in virulence-related genes of Bordetella pertussis isolates from Poland in the period 1959–2013. Eur. J. Clin. Microbiol. Infect. Dis. 2015;34:147–152. doi: 10.1007/s10096-014-2216-6
- Safarchi A, Octavia S, Luu LDW, et al. Better colonisation of newly emerged Bordetella pertussis in the co-infection mouse model study. Vaccine. 2016;34:3967–3971.