
ABSTRACT
Flaviviruses are (re)-emerging RNA viruses strictly dependent on lipid metabolism for infection. In the search for host targeting antivirals, we explored the effect of pharmacological modulation of fatty acid metabolism during flavivirus infection. Considering the central role of acetyl-Coenzyme A carboxylase (ACC) on fatty acid metabolism, we analyzed the effect of three small-molecule ACC inhibitors (PF-05175157, PF-05206574, and PF-06256254) on the infection of medically relevant flaviviruses, namely West Nile virus (WNV), dengue virus, and Zika virus. Treatment with these compounds inhibited the multiplication of the three viruses in cultured cells. PF-05175157 induced a reduction of the viral load in serum and kidney in WNV-infected mice, unveiling its therapeutic potential for the treatment of chronic kidney disease associated with persistent WNV infection. This study constitutes a proof of concept of the reliability of ACC inhibitors to become viable antiviral candidates. These results support the repositioning of metabolic inhibitors as broad-spectrum antivirals.
Introduction
The genus Flavivirus includes 53 closely related species of positive-strand RNA viruses transmitted by vectors (mosquitoes and ticks) [Citation1]. Flaviviruses are responsible for mild-flu like symptoms, neurological syndromes, birth defects, and hemorrhagic fevers. For instance, West Nile virus (WNV) provokes outbreaks of febrile illness, encephalitis, acute flaccid-paralysis and can induce long lasting sequelae and a chronic renal disease associated with persistent infection, dengue virus (DENV) causes about 100 million cases of disease each year including hemorrhagic fevers, and Zika virus (ZIKV) is responsible for birth defects (microcephaly) and neurological syndromes [Citation2–5]. The increase in worldwide travel and trade, global warming, and urbanization are facilitating the colonization of new territories by pathogenic flaviviruses, putting human and animal health at risk. This can be easily exemplified by the emergence of WNV and ZIKV in the Americas from 1999 and 2015, respectively. Despite their clinical relevance, most flaviviruses are still neglected pathogens and there are no specific licensed therapies to combat them. Consequently, there is an urgent need for effective therapies not only against recognized pathogenic flaviviruses, but also potentially suitable against future flaviviral threats.
The advances in the understanding of virus-host interactions have led to the identification of the essential cellular pathways for infection, which has allowed new antiviral approaches directed against host factors. In comparison to direct-acting antivirals that target unique viral factors, host-directed antivirals should be advantageous due to their potential broad spectrum and their theoretical higher genetic barrier to the selection of resistant mutants [Citation6]. Flaviviruses ideally constitute an appropriate objective for this kind of host-directed antiviral discovery that may result in the identification of pan-flaviviral drugs, providing low cost but effective control tools [Citation7–9].
Most viruses, including flaviviruses, are forced to co-opt for specific cellular lipids to complete their life cycles. Due to this fact, lipid metabolism has become an attractive target for host-directed antiviral interventions [Citation10,Citation11]. For instance, a wide variety of viruses are strictly dependent on fatty acid metabolism [Citation12–17]. Flaviviruses also share this dependence on fatty acid metabolism for infection. Fatty acids provide the building blocks for the synthesis of complex lipids that are necessary for flavivirus replication and particle morphogenesis, promote energy production in infected cells, and participate in the immune response [Citation18–23]. A key step within fatty acid metabolism, catalyzed by the acetyl-CoA carboxylase (ACC), is the ATP dependent carboxylation of acetyl-CoA to produce malonyl-CoA [Citation24]. The malonyl-CoA is the essential and rate-limiting substrate for de novo lipogenesis and inhibits the transport of long chain fatty acyl-CoAs across the mitochondrial membrane where they can enter fatty acid oxidation. Therefore, ACC regulates both fatty acid synthesis and oxidation. The central role of ACC in fatty acid metabolism allows this enzyme to be a potential target for the treatment of metabolic diseases and cancer [Citation25]. Despite the strong evidence supporting the role of fatty acid metabolism during viral infections, to our knowledge, the potential of ACC as an antiviral target has not been evaluated in vivo.
In this study, we analyzed the antiviral effect resulting from modulation of fatty acid metabolism through ACC inhibition. Pharmacological blockage of ACC inhibited flavivirus multiplication in cultured cells and reduced viral load in mice. Our results provide experimental evidence to support the hypothesis that ACC is as a druggable target suitable for antiviral intervention.
Materials and methods
Ethics statement
Mouse pharmacokinetic studies were conducted in AAALAC accredited facilities and reviewed and approved by Pfizer Institutional Animal care and Use Committee. All experiments with infectious viruses were conducted in biosafety level 3 facilities and approved by the Ethical Committee of Animal Experimentation of Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA, Madrid, Spain) and by the Division of Animal Protection of the Comunidad de Madrid (PROEX 187/17). Animals were handled in strict accordance with the guidelines of the European Community 86/609/CEE.
Infections
Infectious virus manipulations were performed in biosafety level 3 facilities. The origin of WNV NY99, ZIKV PA259459 and DENV-2 has been described [Citation26]. Infections and virus titrations were carried as described [Citation26] using Vero cells (catalog no. CCL-81; ATTC, Manassas, VA). Unless otherwise specified, Vero cells grown on 24-well culture plates (∼2 × 105 cells/well) were infected with the viruses at a multiplicity of infection (MOI) of 1 plaque forming units (PFU)/cell. Unless otherwise specified, inhibitors were added to cultured cells 1 h after virus inoculation when viral inoculum was replaced by culture medium supplemented with 1% fetal bovine serum. At the end of the treatment (24 h of infection for WNV and ZIKV and 48 h for DENV) virus released to the supernatant of infected cultures was harvested and subsequently titrated. Virus titer was calculated as the number of (PFU)/mL by standard plaque assay using semisolid agarose medium. Antiviral potencies were estimated by calculating the absolute half maximal effective concentration [EC50] from a 4 point dose–response curve including 0, 0.5, 1 and 5 µg/mL of each drug. When the smallest concentration of the compound tested induced a reduction higher than 50%, it was displayed that the EC50 was lower than this concentration. Virucidal assays were performed as described [Citation34]. Briefly, the same amount of WNV (∼ 3 × 108 PFU) was preincubated with the compounds (5 µg/mL) or drug vehicle for 1 h at 37°C in culture medium and then titrated by standard plaque assay to determine the remaining infectivity. For time of addition assays, Vero cells were infected with WNV (MOI of 1 PFU/cell) and treated with the compounds (5 µg/mL) or the equivalent drug vehicle at the desired time points: only 1 h before virus inoculation (drug pre-infection), 1 h after virus inoculation (drug post-infection), 1 h before virus inoculation and during infection (drug pre-infection and post-infection), or left untreated (vehicle). Virus yield in supernatant was determined by plaque assay 24 h after infection.
Inhibitors
(-)-Epicgallocathechin gallate, EGCG (catalog no. 0981 S) was from Extrasynthese (Genay, France). The procedures for the synthesis of PF-05175157 [Citation27], PF-05206574 [Citation28] and PF-06256254 [Citation29] have been previously reported. PF-05175157, PF-05206574 and PF-06256254 were synthesized by Pfizer according to described procedures [Citation27–29]. PF-05175157 is commercially available from Sigma (catalog no. PZ0299). Apparent permeability was assessed in low-efflux MDCKII cells [Citation30]. In vitro potencies of the inhibitors were determined using a radiometric assay [Citation27]. For tissue culture experiments, drugs were suspended in DMSO. Control cells were treated in parallel with the same amount of DMSO (vehicle). Cell viability was estimated in uninfected cells by ATP measurement using Cell Titer Glo luminescent cell viability assay (catalog no. G7579; Promega, Madison, WI). For this purpose, cells were seeded in 96-well plates and were treated with the inhibitors in culture medium supplemented with 1% fetal bovine serum for 24 or 48 h. Cells were lysed by adding equal amounts of Cell Titer Glo reagent (100 µL/well) and gentle rocking (2 min) in an orbital shaker. The amount of ATP in each sample was determined by luminescence measurement using white microplates and a TECAN Genius (Zurich, Switzerland) microplate reader.
Mice
Pharmacokinetics was analyzed after a single oral (p.o.) dose of the compounds (in a 0.5% methyl cellulose suspension) administered to fasted male mice. A dose of 15 mg/kg for PF-05175157 or 100 mg/kg in the case of PF-05206574 and PF-06256254 was analyzed. Antiviral activity in vivo was determined using eight-week-old Swiss albino CD-1 female mice (Envigo, Huntingdon, UK). Animals were treated with PF-05175157 (20 mg/kg) suspended in 1% carboxymethylcellulose by oral gavage (p.o.) twice a day from 1 d before infection with WNV (1 × 104 PFU/mouse intraperitoneally, i.p.) and up to 7 days post-infection. Control mice were treated in parallel with drug vehicle (carboxymethylcellulose). For experiments evaluating the effect of genetic deletion of ACC2 gene on WNV infection, a breeding colony of C57BL/6 ACC2-/- mice was established from two heterozygous Acabtm1Dejs females [Citation31]. Age- and sex-matched eight-week-old ACC2-/- and control wild type (WT) mice were challenged with WNV (1 × 104 PFU/animal, i.p.). Animals were monitored daily and received water and food ad libitum. Animals exhibiting clear signs of disease were anesthetized and sacrificed as were all surviving mice at the end of the experiment.
Lipidomics
Lipid extractions, identification, and quantification by mass spectrometry of Vero cells (106 cells/determination) treated with the inhibitors (5 µg/mL, 24 h) or drug vehicle were performed as described [Citation32].
Virus-like particle (VLP) assay
The amount of VLP released by HeLa3-WNV cells was determined by enzyme-linked immunodot assay [Citation32] using mouse monoclonal anti-WNV envelope protein antibody (catalog no. MAB8150; EMD Millipore, Billerica, MA). To this end, HeLa3-WNV were treated with drug vehicle or ACC inhibitors (5 µg/mL) for 20 h. At this time, culture medium was removed and replaced by fresh medium containing the ACC inhibitors or drug vehicle, and cells were further incubated 4 h more. Culture supernatants containing the VLPs secreted during this 4 h of incubation were then harvested and the amount of VLPs was determined by enzyme-linked immunodot assay.
Immunofluorescence
Immunofluorescence detection of dsRNA using monoclonal antibody J2 (Catalog no. 10010200; Scicons, Szirák, Hungary) in cells infected with WNV (MOI of 1 PFU/cell) and treated with 5 µg/mL of each ACC inhibitor, or drug vehicle, was performed as described [Citation20]. Samples were observed with a confocal laser scanning microscope Leica SPE (Leica, Wetzlar, Germany).
Quantification of viral RNA
RNA was automatically extracted using a QIAcube equipment (QIAGEN GmbH, Hilden, Germany) and QIAamp viral RNA Mini Kit (QIAGEN, catalog no. 52906) for sera or RNeasy Mini Kit (QIAGEN, catalog no. 74106) for tissue homogenates. The content of viral RNA in each sample was analyzed by real-time fluorogenic reverse transcriptase PCR by comparison with previously titrated samples and determined as PFU equivalents/mL [Citation22]. In the case of experiments determining the effect of ACC inhibitors on WNV multiplication in Vero cells, these were initially infected with a MOI of 1 PFU/cell and treated with 5 µg/mL of each ACC inhibitor, or drug vehicle, for 24 h.
Statistical analysis
Statistical analyses were performed using GraphPad PRISM 7 for Windows (GraphPad Software Inc., San Diego, CA). Virus yield were compared using Analysis of the Variance (ANOVA) and t-test applying Bonferroni’s correction for multiple comparisons. Tissue and serum viral loads were analyzed by Mann–Whitney test. Lipidomic data were analyzed using multiple t-test and Sidak-Bonferroni correction for multiple comparisons. Statistically significant differences are indicated in the figures: *P < 0.05, **P < 0.01, ***P < 0.001. The number of biological replicates or animals analyzed in each group is denoted by n in the figure legends. Unless otherwise specified data are presented as the mean ± standard deviation (SD).
Results
Antiviral activity of ACC inhibitors against medically relevant flaviviruses
A series of spirocyclic ketone-containing ACC inhibitors was selected, namely PF-05175157 [Citation27], PF-05206574 [Citation28], and PF-06256254 [Citation29] (A). All three compounds exhibited good potencies against the two isoforms of the enzyme (ACC1 and ACC2) from human and rat (half-maximal inhibitory concentration [IC50] = 10–27 nM). The apparent permeability of the compounds (Papp = 7 × 106–24 × 106 cm/s) supported a high exposure in cellular systems (). The cytotoxicity of the compounds was evaluated by cellular ATP determination (B). A dose-dependent reduction of cell viability was observed at high drug concentrations after 24 and 48 h of treatment (half-maximal cytotoxic concentration [CC50] > 500 µM and > 200 µM respectively) (). Given the good potency, high exposure and low cytotoxicity, the antiviral effect of the ACC inhibitors on the infection of three medically relevant flaviviruses (WNV, ZIKV, and DENV) was assayed (C). Based on the different growth kinetics of the viruses, the antiviral activity was assessed after 24 h of infection for WNV and ZIKV and after 48 h of infection for DENV [Citation26]. The three compounds significantly reduced the virus yield in a dose-dependent manner (C) and exhibited similar antiviral potencies (half maximal effective concentration [EC50] in the low µM range) (). To evaluate the balance between efficacy and safety of the drugs, the relation between cytotoxicity and antiviral activity was assessed by calculating the selectivity index (SI) of each compound against WNV, ZIKV, and DENV. In all cases, SI evidenced a good relation between efficacy and safety of the drugs (SI > 100) although some variation was observed among viruses and compounds (). Overall, these results suggest the broad spectrum antiviral effect of the ACC inhibitors and support that this series of compounds could be good lead candidates for additional development.
Figure 1. ACC inhibitors reduce flavivirus multiplication at non-cytotoxic concentrations. (A) Chemical structure of ACC inhibitors PF-05175157, PF-05206574 and PF-06256254. (B) Effect of ACC inhibitors on cell viability. Vero cells were treated for 24 or 48 h with PF-05175157, PF-05206574 or PF-06256254 and cell viability was determined by measuring cell ATP concentrations (n = 4). (C) Inhibition of flavivirus multiplication by ACC inhibitors. Vero cells were infected with the flaviviruses WNV, ZIKV or DENV (MOI of 1 PFU/cell) and 1 h after virus addition, viral inoculum was removed and culture medium containing the various concentrations of the compounds was added. The virus yield was determined as the viral titer in infected supernatants after 24 h (WNV and ZIKV) or 48 h (DENV) of infection (n = 3). Two tailed Student’s t-test P values between Vehicle and each compound concentration are Bonferroni-corrected for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001. Data represent the means ± SDs.
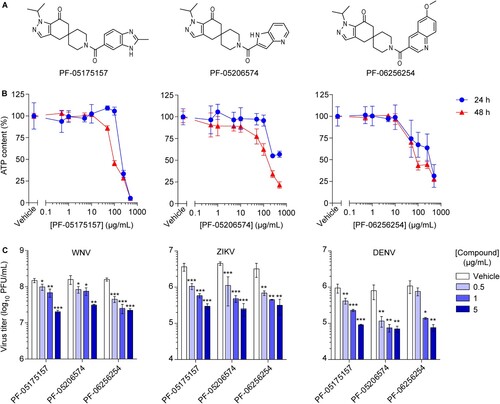
Table 1. Chemical properties and antiviral activities of ACC inhibitors
Preclinical pharmacokinetics of the ACC inhibitors
Considering that the mouse model is commonly used for flavivirus research, the pharmacokinetic of the ACC inhibitors was assessed in this species. The mean plasma concentration-time curves after oral administration (p.o.) of the inhibitors are illustrated in A and the preclinical pharmacokinetics properties are summarized in . Following oral administration, the compounds were rapidly absorbed (time to reach the maximum plasma-concentration [Tmax] = 0.383–1.67 h). All three compounds displayed good oral systemic exposure (area under the plasma-concentration curve [AUCExp] = 2170–3700 ng×h/mL). Compared to PF-05175157, a higher dose was required for PF-05206574 and PF-06256254 to achieve total plasma concentrations (maximum-plasma concentration [Cmax] = 760–1070 ng/mL) that were within the required range for the antiviral activity ( B and ). Accordingly, the pharmacokinetics of the compounds supported their potential antiviral activity in vivo.
Figure 2. Inhibition of ACC reduces viral load in mice. (A) Total plasma level curves of the compounds after a single oral dose (p.o.) of 15 mg/kg for PF-05175157 or 100 mg/kg in the case of PF-05206574 and PF-06256254. Compounds were administered to fasted male mice by oral gavage (n = 3). Data represent the means ± SDs. (B-D) Antiviral activity of PF-05175157 against WNV in mice. (B) Experimental design. Eight-week-old female CD-1 mice were treated with 20 mg/kg of PF-05175157 twice a day p.o. starting 1 d before infection. Animals were infected by the i.p. route with 1 × 104 PFU/mouse of WNV. (C and D) Viral burden in (C) serum and (D) tissues (brain, spleen, liver, kidney and lungs) was determined by quantitative RT-PCR at the indicated times post-infection. Symbols represent individual mice pooled from two independent experiments (n = 8–12). Two-tailed Mann Whitney test P values are indicated in (B) *P = 0.0206, ***P < 0.0001 and (C) *P = 0.0266. (E-G) Infection with WNV in ACC2-deficient mice. (E) Experimental design. Eight-week-old sex-matched WT and ACC2-/- mice were infected by the i.p. route with 1 × 104 PFU/mouse of WNV (n = 10 for WT; n = 9 for ACC2-/-). Viral load in serum (F) or organs (brain, liver, kidney and lung) (G) of infected WT or ACC2-/- mice was determined by quantitative RT-PCR at the indicated times post-infection (n = 9–10 for WT; n = 8–9 for ACC2-/-). Symbols represent individual mice. Two-tailed Mann Whitney test P values is indicated in (E) *P = 0.0244.
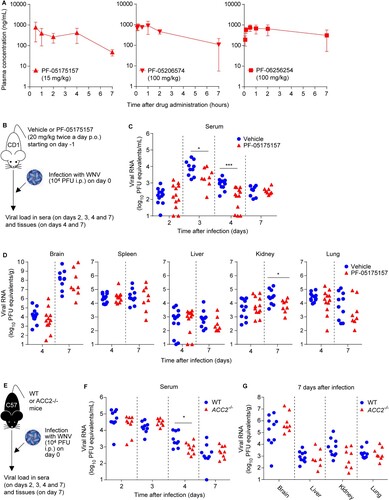
Table 2. Preclinical pharmacokinetics of the ACC inhibitors.
Inhibition of ACC reduces viral load in mice
Based on the safety, potency, and pharmacokinetics attributes of PF-05175157 in mice () and humans [Citation27], this compound was advanced for infection studies in vivo. For this purpose, WNV was selected as a model flavivirus because the murine model of WNV infection is more amenable and resembles clinical signs and pathology more accurately than DENV and ZIKV. Animals were treated with PF-05175157 (20 mg/kg) or drug vehicle twice a day p.o. and viral burden in serum and tissues was analyzed by quantitative RT–PCR up to 7 days after infection (B). When compared to control animals, mice treated with PF-05175157 displayed a significant reduction of viral load in plasma at 3 and 4 days after infection (C). Regarding the viral burden in other tissues, a significant reduction in the amount of viral RNA in the kidney and a tendency for a reduction in the amount of viral RNA in the lung from mice treated with PF-05175157 was noticed at 7 days post-infection (D). Because PF-05175157 inhibits both ACC1 and ACC2 (), the usage of genetic models deficient on ACC1 or ACC2 was considered in order to dissect the role of ACC isoforms on flavivirus infection. Consequently, control wild type (WT) and ACC2 deficient (ACC2-/-) mice [Citation31] were infected with WNV and the viral burden was compared (E). Genetic depletion of ACC2 caused a significant reduction of the viral load at 4 days post-infection (F). However, no significant differences were observed in the organs analyzed (brain, liver, kidney and lung) at 7 days after infection, although a tendency for a reduction in viral burden in the kidney of infected animals was observed (G). These results suggested that the genetic depletion of ACC2 only partially reproduced the effect observed with PF-05175157, indicating that inhibition of ACC1 should be also necessary to achieve the effect observed with PF-05175157. Unfortunately, ACC1 deficiency is embryonically lethal, impairing the use of genetically engineered animal models to evaluate the role of this isoenzyme [Citation33]. Overall, these results reveal the involvement of ACC in flavivirus replication in mouse models and prove the in vivo antiviral activity of ACC inhibitors such as PF-05175157.
Mechanism of inhibition of flavivirus multiplication by ACC inhibitors
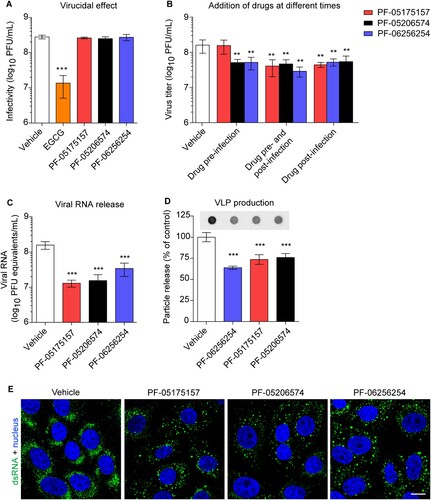
The influence of ACC inhibitors on cellular lipids were investigated. For this end, lipidomics analyses of cells treated with the compounds were performed (). A dose of the ACC inhibitors (5 µg/mL) that exhibited negligible cytotoxicity (B) but significantly inhibited flavivirus replication (C) was selected for these analyses. As expected, the ACC inhibitors significantly modified host lipid landscape, mainly decreasing a wide variety of lipids such as sphingolipids, glycerophospholipids, lysolipids, plasmalogens, and neutral lipids (A and B). To identify the step of the flavivirus life cycle that was impaired by this alteration of lipid metabolism, WNV was selected again as a model. To exclude the possibility that the reduction of flavivirus production was caused as a result of a virucidal effect of the drug, the direct effect of the ACC inhibitors on the infectivity of WNV was evaluated. The same amount of plaque forming units (PFU) was preincubated with the compounds and then titrated to determine the remaining infectivity. In contrast to (-)-Epicgallocathechin gallate (EGCG), included as a positive control of a drug with virucidal effect [Citation34], none of the ACC inhibitors assayed reduced WNV infectivity (A). Consequently, time of addition experiments of ACC inhibitors showed that PF-05175157 did not affect WNV infection when added only prior to virus addition but reduced viral production when maintained after virus inoculation (B). PF-05206574 and PF-06256254 reduced the infection when added prior to virus inoculation in a similar manner than when added after virus inoculation, suggesting subtle differences with PF-05175157 that could produce this long lasting effect. The ACC inhibitors significantly reduced the production of genome containing particles thus confirming the reduction in the production of progeny virions (C). Flavivirus replication and morphogenesis are strictly connected to lipid metabolism [Citation11]. To evaluate if the alteration of lipid metabolism was also related to an impairment of viral morphogenesis, a cell line that expresses WNV structural proteins and constitutively secretes virus like particles (VLPs) without the need of viral replication was used [Citation32,Citation35]. Immunoblot analysis of the amount of released VLPs indicated that the compounds significantly reduced the production of VLPs (D). Flavivirus replication and morphogenesis take place coupled into specialized virus-induced membrane structures derived from the endoplasmic reticulum [Citation36]. The visualization of flavivirus replication complexes by means of immunofluorescence detection of double-stranded RNA (dsRNA) intermediates confirmed that treatment with the ACC inhibitors had also an impact on the development of WNV replication complexes (E). Taken together, these results suggest that the modification of lipid metabolism exerted by ACC inhibitors impaired flavivirus biogenesis by affecting both replication and particle morphogenesis.
Figure 3. Impact of ACC inhibitors on host lipids. (A) Changes in the lipidome of Vero cells after 24 h of treatment with PF-05175157, PF-05206574 or PF-062562545 (5 µg/mL). Fold change in lipid levels were calculated as the log2 (Treated/Vehicle) for each lipid class. Data are expressed as the means ± SDs (n = 3 - 4). Two tailed Student’s t-test P values are Sidak-Bonferroni-corrected for multiple comparisons (*P <0.05, **P <0.01, ***P <0.0001). (B) Heatmap displaying the effect of PF-05175157, PF-05206574 or PF-06256254 on the individual lipid species analyzed. Cells were treated as described in A. Red and blue denote an increase or a decrease in lipid species, respectively. Data correspond to the mean of 3–4 repeats (n = 3-4). Sphingolipids (Cer, ceramide; SM, sphingomyelin), glycerophospholipids (PC, phosphatidylcholine; PE, phosphatidylethanolamine, and PS, phosphatidylserine), diacylglycerol (DAG), and triacylglycerol (TAG) were annotated using “C followed by the total fatty acyl chain length:total number of unsaturated bonds” and the “lipid subclass”. If the sphingoid base residue was dihydrosphingosine, the name contained a “DH” prefix. For glucosylceramides and lactosylceramides a “Gluc” or “Lac” prefix was respectively added to indicate the presence of the sugar moiety. Plasmalogens and lysophospholipids were annotated as described above, except that a “p” or “L” prefix was included, respectively.
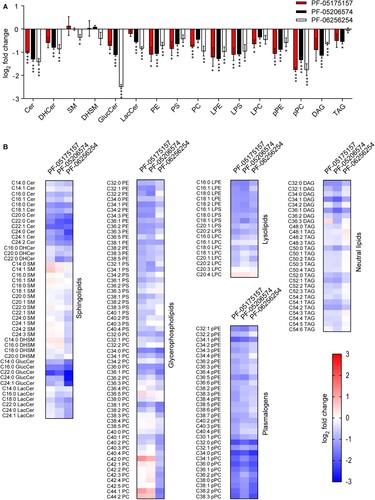
Figure 4. Mechanism of inhibition of WNV multiplication by ACC inhibitors. (A) Lack of virucidal effect of PF-05175157, PF-05206574 or PF-06256254. The same amount of WNV (∼ 3 × 108 PFU) was treated with each compound (5 µg/mL) or drug vehicle for 1 h at 37°C in culture medium and the infectivity in each sample was determined by plaque assay. EGCG was included in the experiments as a positive control of a compound with virucidal activity (n = 3). (B) Time of addition experiments of ACC inhibitors. Vero cells were infected with WNV (MOI of 1 PFU/cell) and treated with PF-05175157, PF-05206574 or PF-06256254 (5 µg/mL) or drug vehicle only 1 h before virus inoculation (drug pre-infection), 1 h after virus inoculation (drug post-infection), 1 h before virus inoculation and during infection (drug pre-infection and post-infection), or left untreated (vehicle). Virus yield in supernatant was determined 24 h after infection by plaque assay (n = 3). (C) Determination by quantitative RT-PCR of genome containing particles released in cell cultures infected with WNV (MOI of 1 PFU/cell) and treated with ACC inhibitors (5 µg/mL) from 1 h after virus inoculation. Viral RNA was quantified 24 h after infection. (D) Production of WNV VLPs by HeLa cells stably transfected with a plasmid that expresses WNV prM and E proteins. HeLa3-WNV cells stably expressing WNV prM and E proteins were treated with ACC inhibitors (5 µg/mL) or drug vehicle and the amount of VLPs released to the culture medium was analyzed by enzyme-linked immunodot assay using a monoclonal antibody against WNV envelope protein as described in Material and methods. The graph displays the quantification of the amount of VLPs released (n = 4). (E) Detection of dsRNA in Vero cells infected with WNV (MOI of 1 PFU/cell) and treated with ACC inhibitors (5 µg/mL) from 1 h after virus inoculation. Cells were fixed and processed for immunofluorescence 24 h post-infection. Scale bar, 10 µm. In (A, B, C, and D) two tailed Student’s t-test P values between Vehicle and each compound and each treatment Bonferroni-corrected for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001. Data represent the means ± SDs.
Discussion
The current progress in deciphering the puzzle of virus-host interactions is helping to develop alternative antiviral strategies. In this context, the close relation between flaviviruses and cellular lipids could drive to new therapeutic opportunities [Citation11,Citation37]. Although the recent increase on the information relative to the involvement of lipids in flavivirus infection [Citation38–40], there are limited data supporting the role of host lipids during flavivirus infection in vivo, and only the relevance of cholesterol and sphingolipids has been demonstrated in animal models [Citation41–44].
Taking advantage of a series of small-molecule ACC inhibitors recently developed to treat human disorders, we have investigated the significance of fatty acid metabolism in the infection of three different flaviviruses (WNV, DENV, and ZIKV) in cell culture. The studies were further extended in vivo using WNV as a model. The ACC inhibitors here used (PF-05175157, PF-05206574, and PF-06256254) displayed excellent in vitro and in cell culture properties (high potency and permeability, low cytotoxicity and good efficacy) and exhibited adequate pharmacokinetic parameters in animal models (like absorption or exposure). Our results showed that the ACC inhibitors reduced the multiplication of WNV, DENV, and ZIKV in cultured cells, which is compatible with the predicted broad spectrum of host targeting antivirals. Among the compounds tested, PF-05175157 has undergone multiple clinical trials in healthy volunteers (NCT01433380), and for the treatment of diabetes mellitus (NCT01792635). Consequently, PF-05175157 was selected as a lead compound for animal experiments. PF-05175157 reduced the peak of viremia in WNV-infected mice, supporting that ACC is a valuable target for antiviral intervention. This effect could be useful to reduce viral load, helping to control the infection or to diminish flavivirus transmission. In addition, PF-05175157 also reduced the viral burden in the kidney of infected mice. Importantly, WNV can induce persistent renal infection in humans and rodents causing chronic kidney disease [Citation5,Citation45], which means that the virus can be recovered from the urine of infected patients not only during acute infection, but also up to several years after infection [Citation46–50]. Therefore, ACC inhibitors like PF-05175157 could turn into valuable tools for the prevention and treatment of renal complications associated to persistent WNV infection. Regarding the safety concerns for the therapeutic utilization of lipid metabolism inhibitors, the clinical success of the pharmacological manipulation of lipid metabolism with widely used drugs like statins or metformin has to be taken into consideration [Citation51,Citation52]. Moreover, metformin targets the adenosine monophosphate-activated protein (AMPK), which is a major regulator of the ACC activity, further supporting the safety of pharmacological manipulation of fatty acid metabolism as a potentially suitable therapeutic strategy [Citation52]. Although treatment with ACC inhibitors failed to completely control viral infection, the reduction of viral load in serum and kidney of infected mice could contribute to the development of novel therapies to combat certain aspects of WNV infection.
The treatment with the ACC inhibitors elicited a major impact on cell lipid metabolism, as determined by lipidomics of treated cells. This effect was similar to the one described for another structurally-related ACC inhibitor [Citation53]. A wide variety of lipids were reduced upon ACC inhibition. These included lipids with proven roles in flavivirus replication complex organization and morphogenesis like ceramides (Cer), glycerophospholipids, lysolipids, and neutral lipids [Citation21,Citation32,Citation54–57]. Our results showed that treatment with ACC inhibitors had a direct impact on WNV infection by affecting both genome replication and particle assembly, two coupled processes strictly dependent on specific lipids necessary for membrane curvature, replication complex assembly, and virion envelopment [Citation11,Citation58]. Accordingly, the modulation of lipid fatty acid synthesis, impairs WNV multiplication [Citation20,Citation22]. The mechanism of inhibition of flavivirus multiplication of ACC inhibitors tested in this study was similar to the effect observed with other ACC inhibitors such as 5-(Tetradecyloxy)-2-furoic acid (TOFA) or 3,3,14,14-Tetramethylhexadecanedioic acid (MEDICA 16) [Citation22]. Additionally, mouse experiments suggested that the inhibition of ACC1 (alone or together with ACC2) could be responsible for the antiviral effect of PF-05175157 in mouse models. This is consistent with the view that ACC1 has been mainly associated with lipogenesis, whereas ACC2 has been related to the regulation of fatty acid oxidation [Citation24].
In summary, our results indicate that the manipulation of fatty acid metabolism via inhibition of ACC impairs flavivirus infection in cell culture and reduces WNV infection in animal models. Understanding the metabolic connections of viral infections opens new therapeutic opportunities using this type of inhibitors that could be ideally combined with other classical antiviral drugs such as nucleoside analogs to develop effective combinatorial therapies. Although these results provide a proof of concept, it has to be considered that further work directed to the optimization of dose and route is required to find the appropriate window of therapeutic efficacy. In addition, further chemical modifications of the compounds could contribute to maximize their antiviral effect.
Acknowledgements
We thank Margarita Calvo (Department of Biotechnology, INIA, Madrid, Spain) and Alex García (Department of Biomedicinal Chemistry, IQAC-CSIC, Barcelona, Spain) for technical assistance.
Conceptualization W.P.E., K.H., A.F.E-K, G.K., J.C.-S and M.A.M.-A; Methodology, P.R.-I, K.H., D.E.J., A.G.-A.; Investigation, N.J.O., A.-B.B., W.P.E., K.H., A.F.E-K, E.E.-R., A.L.-L., M.A.M-A.; Writing – Original Draft, M.A.M.-A.; Writing – Review & Editing, W.P.E., K. H, G.K., A.F.E-K,, A.L.-L., J.-C.S.; Funding Acquisition, F.S., J.C.-S. and M.A.M.-A.
Disclosure statement
W.P.E. is an employee and share holder of Pfizer Inc. K.H., A. E.-K. and G.K. were previously employed by Pfizer Inc.
Additional information
Funding
Related Research Data
References
- Simmonds P, Becher P, Bukh J, et al. ICTV virus Taxonomy Profile: Flaviviridae. J Gen Virol. 2017 Jan;98(1):2–3. PubMed PMID: 28218572; PubMed Central PMCID: PMC5370391. eng. doi: 10.1099/jgv.0.000672
- Martin-Acebes MA, Saiz JC. West Nile virus: A re-emerging pathogen revisited. World J Virol. 2012 Apr 12;1(2):51–70. PubMed PMID: 24175211; PubMed Central PMCID: PMC3782267. Eng. doi: 10.5501/wjv.v1.i2.51
- Saiz JC, Vazquez-Calvo A, Blazquez AB, et al. Zika virus: the Latest Newcomer. Front Microbiol. 2016;7(496). PubMed PMID: 27148186; PubMed Central PMCID: PMC4835484. eng. doi: 10.3389/fmicb.2016.00496
- Guzman MG, Dengue HE. Dengue. Lancet. 2015 Jan 31;385(9966):453–465. PubMed PMID: 25230594; eng. doi: 10.1016/S0140-6736(14)60572-9
- Barzon L, Pacenti M, Palu G. West Nile virus and kidney disease. Expert Rev Anti Infect Ther. 2013 May;11(5):479–487. PubMed PMID: 23627854; eng. doi: 10.1586/eri.13.34
- Kaufmann SHE, Dorhoi A, Hotchkiss RS, et al. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2018 Jan;17(1):35–56. PubMed PMID: 28935918; eng. doi: 10.1038/nrd.2017.162
- Boldescu V, Behnam MAM, Vasilakis N, et al. Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat Rev Drug Discov. 2017 Aug;16(8):565–586. PubMed PMID: 28473729; PubMed Central PMCID: PMC5925760. eng. doi: 10.1038/nrd.2017.33
- Krishnan MN, Garcia-Blanco MA. Targeting host factors to treat West Nile and dengue viral infections. Viruses. 2014 Feb 10;6(2):683–708. PubMed PMID: 24517970; PubMed Central PMCID: PMC3939478. eng. doi: 10.3390/v6020683
- Saiz JC, Martin-Acebes MA. The Race To find antivirals for Zika virus. Antimicrob Agents Chemother. 2017 Jun;61(6). PubMed PMID: 28348160; PubMed Central PMCID: PMC5444186. eng. doi:10.1128/AAC.00411-17.
- Munger J, Bennett BD, Parikh A, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008 Oct;26(10):1179–1186. PubMed PMID: 18820684; PubMed Central PMCID: PMC2825756. eng. doi: 10.1038/nbt.1500
- Martin-Acebes MA, Vazquez-Calvo A, Saiz JC. Lipids and flaviviruses, present and future perspectives for the control of dengue, Zika, and West Nile viruses. Prog Lipid Res. 2016 Oct;64:123–137. PubMed PMID: 27702593; eng. doi: 10.1016/j.plipres.2016.09.005
- Spencer CM, Schafer XL, Moorman NJ, et al. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J Virol. 2011 Jun;85(12):5814–5824. PubMed PMID: 21471234; PubMed Central PMCID: PMC3126312. eng. doi: 10.1128/JVI.02630-10
- Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014 Mar;10(3):e1004021. PubMed PMID: 24651651; PubMed Central PMCID: PMC3961357. eng. doi: 10.1371/journal.ppat.1004021
- Nchoutmboube JA, Viktorova EG, Scott AJ, et al. Increased long chain acyl-Coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLoS Pathog. 2013;9(6):e1003401. PubMed PMID: 23762027; PubMed Central PMCID: PMC3675155. eng. doi: 10.1371/journal.ppat.1003401
- Koutsoudakis G, Romero-Brey I, Berger C, et al. Soraphen A: A broad-spectrum antiviral natural product with potent anti-hepatitis C virus activity. J Hepatol. 2015 Oct;63(4):813–821. PubMed PMID: 26070407; eng. doi: 10.1016/j.jhep.2015.06.002
- Lyn RK, Singaravelu R, Kargman S, et al. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci Rep. 2014 Apr 1;4:4549. PubMed PMID: 25008545; PubMed Central PMCID: PMC4091094. eng. doi: 10.1038/srep04549
- Fleta-Soriano E, Smutna K, Martinez JP, et al. The Myxobacterial Metabolite Soraphen A inhibits HIV-1 by Reducing virus production and Altering virion Composition. Antimicrob Agents Chemother. 2017 Aug;61(8). PubMed PMID: 28533249; PubMed Central PMCID: PMC5527598. eng. doi: 10.1128/AAC.00739-17
- Heaton NS, Perera R, Berger KL, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A.. 2010 Oct 5;107(40):17345–17350. PubMed PMID: 20855599; PubMed Central PMCID: PMC2951450. eng. doi: 10.1073/pnas.1010811107
- Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010 Nov 18;8(5):422–432. PubMed PMID: 21075353; PubMed Central PMCID: PMC3026642. eng. doi: 10.1016/j.chom.2010.10.006
- Martin-Acebes MA, Blazquez AB, de Oya NJ, et al. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PloS one. 2011;6(9):e24970. PubMed PMID: 21949814; PubMed Central PMCID: PMC3176790. eng. doi: 10.1371/journal.pone.0024970
- Perera R, Riley C, Isaac G, et al. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012 Mar;8(3):e1002584. PubMed PMID: 22457619; PubMed Central PMCID: PMC3310792. eng. doi: 10.1371/journal.ppat.1002584
- Merino-Ramos T, Vazquez-Calvo A, Casas J, et al. Modification of the host cell lipid metabolism induced by Hypolipidemic drugs targeting the acetyl Coenzyme A carboxylase impairs West Nile virus replication. Antimicrob Agents Chemother. 2015 Jan;60(1):307–315. PubMed PMID: 26503654; PubMed Central PMCID: PMC4704205. eng. doi: 10.1128/AAC.01578-15
- Kao YT, Chang BL, Liang JJ, et al. Japanese encephalitis virus nonstructural protein NS5 interacts with mitochondrial trifunctional protein and impairs fatty acid beta-oxidation. PLoS Pathog. 2015 Mar;11(3):e1004750. PubMed PMID: 25816318; PubMed Central PMCID: PMC4376648. eng. doi: 10.1371/journal.ppat.1004750
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253–272. PubMed PMID: 18598135; eng. doi: 10.1146/annurev.nutr.28.061807.155434
- Tong L, Harwood HJ, Jr. Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. J Cell Biochem. 2006 Dec 15;99(6):1476–1488. PubMed PMID: 16983687; PubMed Central PMCID: PMC3837461. eng. doi: 10.1002/jcb.21077
- Jimenez de Oya N, Blazquez AB, Casas J, et al. Direct Activation of adenosine monophosphate-activated protein Kinase (AMPK) by PF-06409577 inhibits flavivirus infection through modification of host-cell lipid metabolism. Antimicrob Agents Chemother. 2018 Apr 30. PubMed PMID: 29712653; eng. doi:10.1128/AAC.00360-18.
- Griffith DA, Kung DW, Esler WP, et al. Decreasing the rate of metabolic ketone reduction in the discovery of a clinical acetyl-CoA carboxylase inhibitor for the treatment of diabetes. J Med Chem. 2014 Dec 26;57(24):10512–10526. PubMed PMID: 25423286; PubMed Central PMCID: PMC4281100. eng. doi: 10.1021/jm5016022
- Bagley SW, Griffith DA, Kung DW. inventors; Pfizer Inc., assignee. N1-pyrazolospiroketone acetyl-Coa carboxylase inhibitors. US 2011111046. May 2011.
- Dow LR, Edmonds DJ, Griffith DA, et al. inventors; Pfizer Inc., assignee. Substituted acetyl-CoA carboxylase inhibitors. US 2012270893. Oct 2012.
- Di L, Whitney-Pickett C, Umland JP, et al. Development of a new permeability assay using low-efflux MDCKII cells. J Pharm Sci. 2011 Nov;100(11):4974–4985. PubMed PMID: 21766308; eng. doi: 10.1002/jps.22674
- Hoehn KL, Turner N, Swarbrick MM, et al. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. 2010 Jan;11(1):70–76. PubMed PMID: 20074529; PubMed Central PMCID: PMC2824926. eng. doi: 10.1016/j.cmet.2009.11.008
- Martin-Acebes MA, Merino-Ramos T, Blazquez AB, et al. The composition of west Nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J Virol. 2014 Oct 15;88(20):12041–12054. PubMed PMID: 25122799; eng. doi: 10.1128/JVI.02061-14
- Abu-Elheiga L, Matzuk MM, Kordari P, et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl. Acad. Sci. U.S.A.. 2005 Aug 23;102(34):12011–12016. PubMed PMID: 16103361; PubMed Central PMCID: PMC1189351. eng. doi: 10.1073/pnas.0505714102
- Vazquez-Calvo A, de Oya N J, Martin-Acebes MA, et al. Antiviral properties of the Natural Polyphenols Delphinidin and Epigallocatechin gallate against the flaviviruses West Nile virus, Zika virus, and dengue virus. Front Microbiol. 2017;8:1314. PubMed PMID: 28744282; PubMed Central PMCID: PMC5504193. eng. doi: 10.3389/fmicb.2017.01314
- Merino-Ramos T, Blazquez AB, Escribano-Romero E, et al. Protection of a single dose West Nile virus Recombinant Subviral particle Vaccine against Lineage 1 or 2 Strains and analysis of the Cross-Reactivity with Usutu virus. PloS one. 2014;9(9):e108056. PubMed PMID: 25229345; Eng. doi: 10.1371/journal.pone.0108056
- Apte-Sengupta S, Sirohi D, Kuhn RJ. Coupling of replication and assembly in flaviviruses. Curr Opin Virol. 2014 Dec;9:134–142. PubMed PMID: 25462445; PubMed Central PMCID: PMC4268268. eng. doi: 10.1016/j.coviro.2014.09.020
- Villareal VA, Rodgers MA, Costello DA, et al. Targeting host lipid synthesis and metabolism to inhibit dengue and hepatitis C viruses. Antiviral Res. 2015 Dec;124:110–121. PubMed PMID: 26526588; PubMed Central PMCID: PMC4699661. eng. doi: 10.1016/j.antiviral.2015.10.013
- Zhang J, Lan Y, Sanyal S. Modulation of lipid Droplet metabolism-A potential target for therapeutic intervention in Flaviviridae infections. Front Microbiol. 2017;8:2286. PubMed PMID: 29234310; PubMed Central PMCID: PMC5712332. eng. doi: 10.3389/fmicb.2017.02286
- Pombo JP, Sanyal S. Perturbation of Intracellular cholesterol and fatty acid Homeostasis during flavivirus infections. Front Immunol. 2018;9:1276. PubMed PMID: 29915602; PubMed Central PMCID: PMC5994796. eng. doi: 10.3389/fimmu.2018.01276
- Leier HC, Messer WB, Tafesse FG. Lipids and pathogenic flaviviruses: An intimate union. PLoS Pathog. 2018 May;14(5):e1006952. PubMed PMID: 29746606; PubMed Central PMCID: PMC5944919. eng. doi: 10.1371/journal.ppat.1006952
- Martinez-Gutierrez M, Correa-Londono LA, Castellanos JE, et al. Lovastatin delays infection and increases survival rates in AG129 mice infected with dengue virus serotype 2. PloS one. 2014;9(2):e87412. PubMed PMID: 24586275; PubMed Central PMCID: PMC3931612. eng. doi: 10.1371/journal.pone.0087412
- Li C, Deng YQ, Wang S, et al. 25-Hydroxycholesterol Protects host against Zika virus infection and Its associated microcephaly in a mouse model. Immunity. 2017 Mar 21;46(3):446–456. PubMed PMID: 28314593; PubMed Central PMCID: PMC5957489. eng. doi: 10.1016/j.immuni.2017.02.012
- Martin-Acebes MA, Gabande-Rodriguez E, Garcia-Cabrero AM, et al. Host sphingomyelin increases West Nile virus infection in vivo. J Lipid Res. 2016 Mar;57(3):422–432. PubMed PMID: 26764042; PubMed Central PMCID: PMC4766991. eng. doi: 10.1194/jlr.M064212
- Taniguchi M, Tasaki T, Ninomiya H, et al. Sphingomyelin generated by sphingomyelin synthase 1 is involved in attachment and infection with Japanese encephalitis virus. Sci Rep. 2016 Nov 28;6(37829). PubMed PMID: 27892528; PubMed Central PMCID: PMC5124946. eng. doi:10.1038/srep37829.
- Saxena V, Xie G, Li B, et al. A hamster-derived West Nile virus isolate induces persistent renal infection in mice. PLoS Negl Trop Dis. 2013;7(6):e2275. PubMed PMID: 23785537; PubMed Central PMCID: PMC3681636. eng. doi: 10.1371/journal.pntd.0002275
- Barzon L, Pacenti M, Franchin E, et al. Excretion of West Nile virus in urine during acute infection. The Journal f Infectious Diseases. 2013 Oct 1;208(7):1086–1092. PubMed PMID: 23821721; eng. doi: 10.1093/infdis/jit290
- Murray KO, Kolodziej S, Ronca SE, et al. Visualization of West Nile virus in urine Sediment using Electron Microscopy and Immunogold up to Nine years Postinfection. Am J Trop Med Hyg. 2017 Dec;97(6):1913–1919. PubMed PMID: 29141749; PubMed Central PMCID: PMC5805063. eng. doi: 10.4269/ajtmh.17-0405
- Nolan MS, Podoll AS, Hause AM, et al. Prevalence of chronic kidney disease and progression of disease over time among patients enrolled in the Houston West Nile virus cohort. PloS one. 2012;7(7):e40374. PubMed PMID: 22792293; PubMed Central PMCID: PMC3391259. eng. doi: 10.1371/journal.pone.0040374
- Ergunay K, Karagul A, Abudalal A, et al. Prospective investigation of the impact of West Nile virus infections in renal diseases. J Med Virol. 2015 Oct;87(10):1625–1632. PubMed PMID: 25965349; eng. doi: 10.1002/jmv.24226
- Murray K, Walker C, Herrington E, et al. Persistent infection with West Nile virus years after initial infection. J Infect Dis. 2015 Jan 1;201(1):2–4. PubMed PMID: 19961306; PubMed Central PMCID: PMC2791189. eng. doi: 10.1086/648731
- Opie LH. Present status of statin therapy. Trends Cardiovasc Med. 2015 Apr;25(3):216–225. PubMed PMID: 25802224; Eng. doi: 10.1016/j.tcm.2014.10.002
- Hardie DG. Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface. 2018 Jan;15(138):20170774. PubMed PMID: 29343628; PubMed Central PMCID: PMC5805978. eng. doi: 10.1098/rsif.2017.0774
- Jones JE, Esler WP, Patel R, et al. Inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) reduces Proliferation and De novo lipogenesis of EGFRvIII human Glioblastoma cells. PloS one. 2017;12(1):e0169566. PubMed PMID: 28081256; PubMed Central PMCID: PMC5231342 ONE policies on sharing data and materials. eng. doi: 10.1371/journal.pone.0169566
- Aktepe TE, Pham H, Mackenzie JM. Differential utilisation of ceramide during replication of the flaviviruses West Nile and dengue virus. Virology. 2015 Jun 26;484:241–250. PubMed PMID: 26122470; Eng. doi: 10.1016/j.virol.2015.06.015
- Liebscher S, Ambrose RL, Aktepe TE, et al. Phospholipase A2 activity during the replication cycle of the flavivirus West Nile virus. PLoS Pathog. 2018 Apr;14(4):e1007029. PubMed PMID: 29709018; PubMed Central PMCID: PMC5945048. eng. doi: 10.1371/journal.ppat.1007029
- Jordan TX, Randall G. Dengue virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J Virol. 2017 Jun 1;91(11). PubMed PMID: 28298606; PubMed Central PMCID: PMC5432877. eng. doi: 10.1128/JVI.02020-16
- Zhang J, Lan Y, Li MY, et al. Flaviviruses Exploit the lipid Droplet protein AUP1 to Trigger Lipophagy and drive virus production. Cell Host Microbe. 2018 Jun 13;23(6):819–831 e5. PubMed PMID: 29902443; eng. doi: 10.1016/j.chom.2018.05.005
- Aktepe TE, Mackenzie JM. Shaping the flavivirus replication complex: it's Curvaceous!. Cell Microbiol. 2018 Jun 22;20:e12884. PubMed PMID: 29933527; eng. doi: 10.1111/cmi.12884