
ABSTRACT
Background: Multidrug-resistant bacteria, especially those with high virulence, are an emerging problem in clinical settings.
Methods: We conducted a multicentre epidemiological and comparative genomic analysis on the evolution, virulence and antimicrobial resistance of carbapenem-resistant Enterobacteriaceae in patients with bacterial liver abscesses from 2012 to 2016.
Results: A total of 477 bacterial isolates were collected. Enterobacteriaceae were the main pathogen (89.3%) with K. pneumoniae (52.4%) predominating followed by Escherichia coli (26.8%). All CRKps (3.2%) were of sequence type (ST) 11 and serotypes K47 or K64, and simultaneously possessed acquired blaKPC-2/blaKPC-5 and blaCTX-M-65 together with the multidrug transporter EmrE. Seven Hv-CRKps (five ST11-K47, two ST11-K64) were confirmed by bacteriological test, neutrophil killing assay and Galleria mellonella infection model. Genomic analysis indicated that the emergence of one ST11-K64 Hv-CRKp strain was related to the acquisition of rmpA/rmpA2 genes and siderophore gene clusters, while ST11-K47 Hv-CRKp lacked these traditional virulence genes. Further complete genome analysis of one ST11-K47 Hv-CRKp strain, R16, showed that it acquired a rare plasmid (pR16-Hv-CRKp1) carrying blaKPC-2, blaSHV-12, blaTEM-1, blaCTX-M-65, rmtB and a predicted virulence gene R16_5486 simultaneously.
Conclusion: The emergence of the ST11-K47/K64 Hv-CRKps, which are simultaneously multidrug-resistant and hypervirulent, requires urgent control measures to be implemented.
Introduction
The increased prevalence of antibiotic resistance among pathogens is nearly at critical levels. In 2016, the World Health Organization (WHO) came up with a pathogen list of the most important resistant bacteria at a global level for which there is an urgent need for new treatments, with carbapenem-resistant Enterobacteriaceae (CRE) classified as a critical priority organism. Infections caused by CRE have emerged worldwide, posing a global public health threat and a formidable challenge to antimicrobial therapy [Citation1,Citation2]. Among these, carbapenem-resistant Klebsiella pneumoniae strains (CRKp) account for roughly 70–90% of clinical CRE infections in the European Union and China [Citation3,Citation4].
Bacterial liver abscess is a relatively common infection caused by a wide variety of bacteria. Before mid-1980s, Escherichia coli was the most common etiological agent of pyogenic liver abscesses (PLA), until a new hypervirulent variant of K. pneumoniae (HvKp) causing invasive PLA was firstly described in Taiwan, and subsequently found worldwide, especially in Asia, with the most common serotypes being K1 and K2 [Citation5,Citation6]. To date, HvKp is considered to surpass E. coli to become the predominant cause of PLA over the past three decades [Citation7]. Although most of the HvKp strains identified to date are susceptible to most antimicrobials, some cases of multidrug-resistant HvKp infections have been reported [Citation8]. Most recently, carbapenem-resistant HvKp (CR-HvKp) isolated from different clinical settings has begun to emerge in China [Citation9,Citation10]. The possibility of enhanced virulence and acquisition of carbapenem resistance in HvKp strains further complicates clinical practice.
Despite the low incidence of Hv-CRKp strains in China, few studies have been performed to investigate the epidemiology of bacterial liver abscesses caused by CRE. In this study, we conducted a multi-centre study to understand the pathogen epidemiology of bacterial liver abscess, and also carried out a comparative genomic analysis focusing on CR-HvKp strains, to better clarify the virulence and antimicrobial resistance mechanisms involved.
Methods
Bacterial strains
Gram-negative bacteria were consecutively collected from patients with clinical- and laboratory-confirmed bacterial liver abscesses, diagnosed based on the Johns Hopkins ABX Guide (https://www.hopkinsguides.com/hopkins/view/Johns_Hopkins_ABX_Guide/540259/all/Hepatic_Abscess) between 2012 and 2016 in 15 centres located in 11 Chinese cities. All organisms were considered clinically significant by local hospital criteria and were isolated from high-quality specimens, including liver tissues collected in surgery or from liver abscess aspirates. All isolates were sent to the central clinical microbiology laboratory of Peking Union Medical College Hospital (PUMCH) for identification confirmation using MALDI-TOF MS (Vitek MS, BioMérieux, France). The Human Research Ethics Committee of PUMCH approved this study (Ethics Approval Number: S-K238).
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing was conducted by broth microdilution method as per the Clinical and Laboratory Standards Institute (CLSI) recommendations [Citation11]. MICs were interpreted following the CLSI M100-S28 guidelines [Citation11]. E. coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and K. pneumoniae ATCC700603 were used as quality controls.
Virulence assay using the string test, neutrophil killing assay, and Galleria mellonella infection model
To identify the hypermucoviscous phenotype of K. pneumoniae, string test was performed as previously described [Citation12]. The result was deemed positive when a viscous string longer than 5 mm could be generated by touching and pulling a single colony upwards with a standard inoculation loop. Neutrophil killing assay (NKA) was also performed to test the virulence potential of K. pneumoniae isolates [Citation13]. Furthermore, we tested the virulence potential of the isolates in G. mellonella larvae weighing approximately 300 mg (purchased from Tianjin Huiyude Biotech Company, Tianjin, China), as previously described. A known hypervirulent Klebsiella pneumoniae strain 1088, a gift from Dr. Zhang R was used as the positive control [Citation9]. A classic K. pneumoniae strain QD110 identified in this study was added as a negative control (see detailed information in Material S3).
Genomic DNA extraction, sequencing, assembly and annotation
To better understand the genetic basis of CRE strains, all CRE (study group) and 12 carbapenem-susceptible Enterobacteriaceae (CSE) (comparative group) were further analyzed for whole-genome sequencing. Single risk factor analysis concerning sex, age, patient source and some more detailed clinical information between CRE and CSE group was performed using fisher's exact test or Mann–Whitney test. Multivariate Logistic Regression analysis was not performed due to the limited sample sizes. TIANamp Bacteria Genomic DNA Kit (TiangenBiotechCo. Ltd., Beijing, China) and Illumina Genome Analyzer 2X technology (Illumina, San Diego, CA, USA) were used for the genomic DNA extraction and shotgun sequencing. Adapters and low-quality sequences were trimmed and filtered, and SPAdes v3.11was used for the de novo assembly of these reads [Citation14]. Whole-genome sequencing of one ST11-K47 Hv-CRKp strain R16 was performed using Pacific Biosciences Sequel System (Pacific Biosciences, Menlo Park, CA, USA). The sequencing reads of other seven CRKps were mapped on the genome of R16 using BWA 0.5.9, and genome coverage, gene coverage and SNPs were analyzed using samtools and bedtools (see detailed information in Supplementary Material S3).
Sequence types (STs) and serotype analysis
STs were determined based on multilocus sequence typing (MLST, http://bigsdb.Pasteur.fr/klebsiella/klebsiella.html, http://mlst.warwick.ac.uk/mlst/dbs/E.coli). It was also re-confirmed using SRST2 based on the Illumina reads [Citation15]. Serotypes were identified based on the whole genome data using Kaptive (https://github.com/katholt/Kaptive) together with the Blast results between the known primer sequences and assembled scaffolds [Citation16].
Single nucleotide polymorphism (SNP) identification and phylogenetic analysis
SNPs were identified by MUMmer using the published complete genomes. Paired-end reads of K. pneumoniae were mapped to the HS11589 genome (NC_016845.1), and paired-end reads of E. coli were mapped to the O157:H7 genome (NC_002695.1) using BWA 0.5.9. SNPs were analyzed using SAMtools 0.1.19. SNPs located in repetitive regions of the reference genome, defined as exact repetitive sequences ≥25 bp in length as identified using RepeatMasker (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker), were excluded. Phylogenetic tree was constructed using the SNPs described in all strains based on the maximum likelihood in MEGA7 [Citation17] (see detailed information in Material S3).
Virulence genes, antimicrobial resistance genes and homologous genes analysis
Virulence genes in K. pneumoniae and E. coli were downloaded from the K. pneumoniae BIGSdb (http://bigsdb.Pasteur.fr/klebsiella/klebsiella.html) and Virulence Factor Database (VFDB), respectively. Antimicrobial resistance genes were downloaded from the files of ResFinder 3.0 in SRST2 [Citation15]. The virulence and antimicrobial resistance genes in our sequenced strains were predicted using SRST2.
Analysis of plasmids from R16
The plasmids of R16 were further annotated based on ExPASy and NCBI NR databases, and the closely related plasmids [Citation18]. The genome structure comparisons among plasmids were performed to analyze the sequence homology. Plasmid circular structure maps were generated with BRIG software. Prophage analysis was conducted by PHAST [Citation19].
Results
Demographic information and pathogen distribution of patients with bacterial liver abscesses
A total of 477 Gram-negative bacteria isolated from 477 patients with clinical- and laboratory-confirmed bacterial liver abscesses, were studied (). Two-hundred and ninety-one patients (61.0%) were males; the mean age was 59.9 ± 13.1 years. Enterobacteriaceae was the main family of pathogens isolated (426, 89.3%), with K. pneumoniae (250, 52.4%) and E. coli (128, 26.8%) being the top two most commonly isolated species. Non-fermenting Gram-negative bacteria comprised 10.3% of the observed pathogens, with A. baumannii (24, 5.0%) and P. aeruginosa (19, 4.0%) being the two most common ().
Figure 1. Geographic distribution of isolates obtained from bacterial liver abscesses from 2012 to 2016, China.
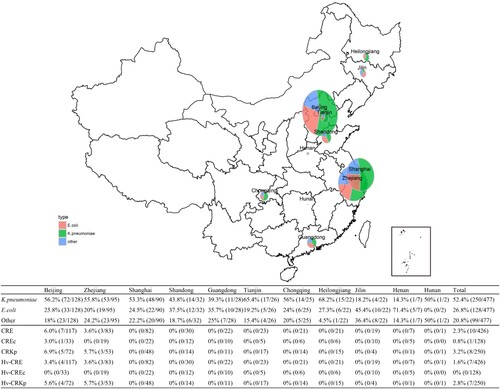
Table 1. The demographic information and pathogen distribution of patients who presented with bacterial liver abscesses from 2012 to 2016, China.
Antimicrobial susceptibility of isolates obtained from bacterial liver abscesses showing the emergence of CRE
Eleven antibiotics were used for antimicrobial susceptibility testing of 426 Enterobacteriaceae isolates. A total of ten (2.4%) CRE (resistant to ertapenem or imipenem) were detected (). K. pneumoniae isolates exhibited a high level of susceptibility to all antimicrobial agents tested, and were most susceptible to ertapenem, imipenem, amikacin, and piperacillin-tazobactam. CRKp accounted for 3.2% (8/250) of all the K. pneumoniae isolates (). Similarly, E. coli isolates also showed a high level of susceptibility to imipenem, ertapenem and amikacin (>90%) with CREc accounting for 0.8% (1/128) (). Moreover, CRKps and CREcs only occured in Beijing and Zhejiang (). Apart from Enterobacteriaceae, A. baumannii showed a high level of resistance to all antimicrobial agents tested with susceptibility rates of <40% ().
Table 2. Antimicrobial susceptibility of K. pneumoniae, E. coli and A. baumannii isolates obtained from bacterial liver abscesses from 2012 to 2016, China.
Clinical information and genetic characteristics of CRE and CSE
CRE (8 K. pneumoniae, 1 E. coli and 1 E. cloacae) were isolated from ten patients aged between 23 and 67 years old in five different hospitals. Four of them had an in-patient history and/or broad-spectrum antibiotic usage within 90 days before infection. They all underwent surgery intervention and at least five days’ treatment with various antibiotics. Carbapenems were used in three of the ten patients, among which the two treated with ertapenem with or without tigecycline got improved and discharged; the one treated with meropenem was not improved based on clinical and bacteriological assessment. Clinical and bacteriological assessment revealed six remission cases with assumed pathogen clearance, four inefficiency or deterioration with two assumed pathogen non-clearance and two pathogens persisting. Nine patients (six improved and three didn't improve) were discharged from hospital after treatment and the remaining one died from infection after 20 days of treatment (Supplementary Table S4).
CSE (7 K. pneumoniae and 5 E. coli) were isolated from ten patients aged between 39 and 67 years old in eight different hospitals. Five of them had an in-patient history and/or broad-spectrum antibiotic usage within 90 days before infection. Imipenem was administered in ten of the patients during their treatment progress. All the patients underwent surgery intervention procedures. Clinical and bacteriological assessment revealed 11 remission cases with eight assumed pathogen clearance, one with assumed not clearance and two with pathogen persisting; one inefficiency or deterioration with pathogen persisting. All but one patient got improved and discharged from hospital (Supplementary Table S4).
Risk factor analysis concerning sex, age, patient source and some more detailed clinical information between CRE and CSE group was performed but none of these factors were considered statistically significant (p > 0.05) (Supplementary Table S5).
MLST genotyping, serotypes and phylogenetic analysis of CRE strains
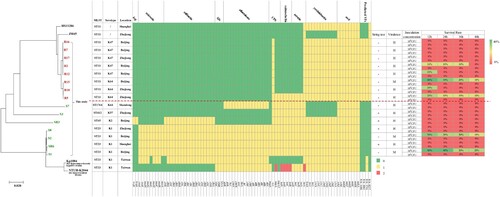
To better understand the genetic basis of CRE strains, the 10 CRE (8 K. pneumoniae, 1 E. coli and 1 E. cloacae) and 12 CSE (7 K. pneumoniae and 5 E. coli), isolated from the same geographic area and same collection period as the 10 CRE, were further analyzed. MLST and serotype results showed that all the CRKps were ST11 and serotypes K47 and K64, whilst most of the CSKps strains belonged to ST23 and serotype K1. We also detected one rare ST1764 CSKp strain. In contrast to the CRE, the CSEcs were of diverse STs (), and one CREc strain belonged to ST88. Moreover, 64 K. pneumoniae and 70 E. coli strains with complete genomes were downloaded from NCBI for phylogenetic tree construction based on whole-genome SNPs (Table S1). As shown in , all CRKps of ST11 were clustered together with ST11 strain HS11286 (NC_016845). ST11-K47 and ST11-K64 strains appear to have evolved separately into two different branches. The four ST23 CSKps were phylogenetically closely related to the K1 hypervirulent strain NTUH-K2044 (NC_012731). The other three CSKp strains clustered with the K2 hypervirulent strain Kp52.145 (NZ_FO834906).
Figure 2. Phylogenetic analysis of K. pneumoniae (A) and E. coli (B) strains. Fifteen K. pneumoniae strains in this study are highlighted in different colours as follows: carmine, ST11; orange, ST23; green, ST412 and ST1764; and blue, ST65. Six E. coli strains in this study are highlighted in carmine. Circles outside the tree indicate the STs and locations of each strain. Different STs and locations are marked with different colours, and strains not from China are coloured grey.
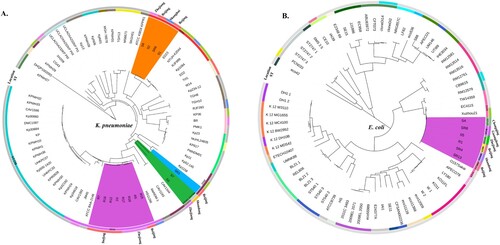
Regarding E. coli, the one CREc and five CSEc strains were clustered in one branch with O157Sakai (Japan 1996 outbreak isolate) and Xuzhou21 (a novel E.coli O157:H7 clone that caused a major hemolytic uremic syndrome outbreak in China) [Citation20]. These results indicate a close relationship among these strains.
Comparative analysis of antibiotic resistance genes
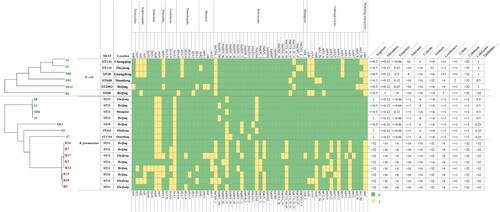
Comparative genomic analysis of antibiotic resistance genes indicated that the CRE strains harboured more types and numbers of drug-resistance genes than those found in CSE. In addition, these CRKps contained almost all classes of genes conferring resistance to aminoglycoside, rifampicin, phenicol, trimethoprim, fosfomycin, macrolide, quinolone, sulphonamide, tetracycline and beta-lactam (). Moreover, carbapenemase genes (blaKPC-2 or blaKPC-5) and extended spectrum β-lactamase (ESBL) genes (blaCTX-M-65) simultaneously existed in CRKp strains. Interestingly, multidrug transporter ErmE that were initially described in E. coli, have now been acquired by all CRKps but not CSKps (). This gene expels positively charged hydrophobic drugs, thereby conferring resistance to a wide range of toxic compounds [Citation21].
Figure 3. Evolutionary relationships, antibiotic resistance genes, distribution and MIC values of K. pneumoniae and E. coli strains. Evolutionary relationships, antimicrobial resistance genes and MICs are shown on the left, middle and right, respectively. Strains are colour coded on the tree (CRE, red; CSE, green). Each row of the heatmap (middle) indicates a strain, and each column represents an antibiotic resistance gene that belongs to the indicated functional clusters shown at the top. The colour intensity shows the copy number of the antibiotic resistance gene.
Virulence phenotype and genetic characteristics of CRE indicate the emergence of ST11-K47 and ST11-K64 Hv-CRKps
Virulence phenotype characterization of CRE
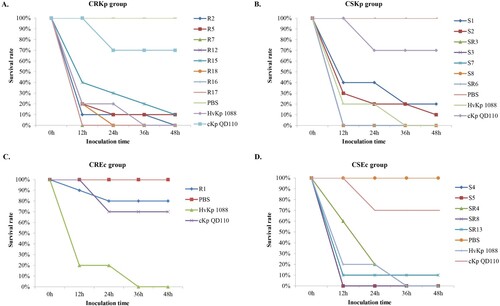
A positive string test was observed in 0/8 of the CRKp and 4/7 of the CSKp isolates. Human NKA results showed that 7/8 of the CRKp and all the CSKp isolates exhibited higher survival rates than strain 1088 (80%) and thus classified as HV. Only one of the CRKp was considered to be MV with a survival rate of 77.9% (). Further animal testing results using a G. mellonella infection model are shown in . Generally, the CRKp and CREc isolates exhibited relatively lower virulence than CSKp and CSEc isolates. However, all five ST11 CRKps isolates with capsular serotype K47 and two ST11-K64 CRKp isolates showed high virulence in the CRKp group. Strains R16, R7 and R17 showed the highest virulence level, exhibiting a survival rate of 0% at 12 h with an inoculation of 106 CFU of bacteria, followed by R2, R12, R18 and R5 (survival rates less than 20% at 12h), comparable to that of strain 1088. In the CSKp group, S7, S3, SR3, S8 and SR6 showed HV with a survival rate of 0% at 12h, most of which were ST23 belonging to the familiar capsular serotypes K1, K2 or K57. For E. coli, four CSEc isolates belonged to the HV group; the remaining one CSEc and one CREc isolates belonged to the low virulence group.
Figure 4. Human neutrophil assays of CRKp and CSKp.
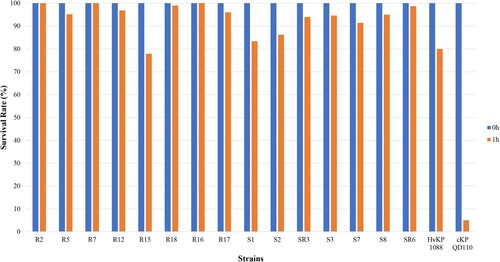
Figure 5. Virulence potential of K. pneumoniae and E. coli strains in a G. mellonella infection model. The effect of 1 × 106CFU of each strain on survival was assessed in G.mellonella. (A) CRKp group. (B) CSKp group. (C) CREc group. (D) CSEc group.
Comparative analysis of virulence genes
A total of 79 K. pneumoniae virulence genes were downloaded from the K. pneumoniae BIGSdb database, and 273 virulence genes of E. coli were downloaded from the VFDB. Comparative virulence gene analysis was conducted among these isolates ( and Figure S1). As shown in , fewer virulence genes were possessed by CRKps than CSKps generally. In the CSKp group, all Hv-CSKps isolates carried the capsular polysaccharide (CPS) regulator genes (rmpA/rmpA2), salmochelin and aerocin siderophore gene clusters, which have been reported to be related to the hypervirulence phenotype of K. pneumoniae [Citation12]. In the CRKp group, only the ST11-K64 Hv-CRKp strain R5 acquired the CPS regulator genes and siderophore genes, which was consistent with its hypervirulence phenotype. The remaining two ST11-K64 and five ST11-K47 Hv-CRKp strains lacked these virulence genes, which implies the likely presence of new virulence genes that need to be discovered.
Figure 6. Virulence gene distribution and virulence assay of 15 K. pneumoniae strains in a G. mellonella infection model. Evolutionary relationships, virulence genes and virulence phenotypes are shown on the left, middle and right, respectively. Two K1 hypervirulent and two ST11 online complete genomes were selected from the NCBI GenBank as references. Our 15 strains are colour coded on the tree (CRKp, red; CSKp, green). Each row of the heatmap (middle) indicates a strain, and each column represents a virulence gene that belongs to the indicated 10 functional clusters shown at the top. The colour intensity shows the copy number of the virulence gene. The virulence assay was conducted using the G. mellonella infection model, and the survival rates of G. mellonella infected with the respective strains are shown on the right.
However, the wzi gene and another two virulence genes, yersinibactin and mrk, were observed in all the Hv-CRKp strains [Citation22]. Therefore, we further conducted a homologous gene analysis against VFDB between the strains with different phenotypes. Four predicted virulence genes (R16_0237, R16_3744, R16_4062, R16_5486) were described only in CRKps compared with our seven CSKps (). Moreover, the predicted virulence gene, R16_3744, was also found in Kp1084 and the hypervirulent strain NTUH-K2044, yet more work need to be done. In contrast, the analysis of virulence genes in E. coli revealed no obvious features (Figure S1 and Table S2).
Complete genome and detailed plasmids analysis of the ST11-K47 Hv-CRKp strain R16
In order to gain further insights into the genetic basis of virulence and drug resistance of Hv-CRKp strains in this study, one ST11-K47 Hv-CRKp strain R16 was further sequenced using PacBio Sequel system. Through genome assembly, we obtained the first complete genome sequence of ST11-K47 Hv-CRKp strain. The bioinformatic analysis providing the general genome information is shown in .
Table 3. Genome information of ST11-K47 Hv-CRKp strain R16.
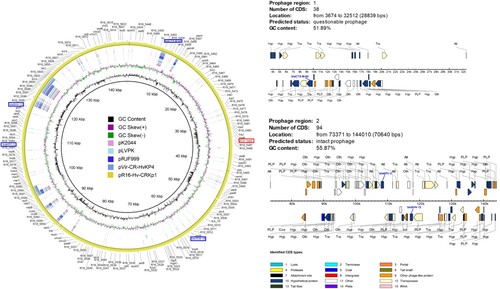
Strain R16 contained three plasmids pR16-Hv-CRKp1, pR16-Hv-CRKp2 and pR16-Hv-CRKp3 (). We found the pR16-Hv-CRKp1 (141 kb) plasmid to be unique as it harboured the carbapenemase genes blaKPC-2, beta-lactamase genes blaCTX-M-65, blaSHV-12 and blaTEM-1, aminoglycoside resistance gene rmtB together with one predicted virulence gene R16_5486, simultaneously. This plasmid is IncFII type and shared 99% identity with p283747-KPC (GenBank MF168406). To the best of our knowledge, this is the first report on this plasmid. Moreover, not much homologous sequences were found between this plasmid and the known virulent plasmids (pK2044 (GenBank AP006726), pLVKP (GenBank AY378100), pRJF999 (GenBank CP014011) and pVir-CR-HvKP4 (GenBank MF437313)) (). Structure analysis revealed two prophages (one intact prophage of 70,640 bp, and one questionable prophage of 28,839 bp) located on pR16-Hv-CRKp1 (). The blaKPC-2 and blaSHV-12 genes were integrated to R16 through the intact 70,640 bp prophage, and blaCTX-M-65 was integrated to R16 through the questionable 288,39 bp prophage, endowing R16 with corresponding drug resistance. The presence of the whole plasmid may simultaneously endow R16 with high virulence due to the predicted virulence gene R16_5486.
Figure 7. Structure of the plasmid pR16-Hv-CRKp1 and corresponding prophage regions. (A) Structural comparison of the plasmid pR16-Hv-CRKp1 and four known virulence plasmids. Alignment of plasmid pK2044, pLVKP, pRJF999 and pVir-CR-HvKP4 are shown as concentric rings. The outermost show the main coding genes of pR16-Hv-CRKP1. Drug resistance genes are highlighted in blue, and the predicted virulence gene is highlighted in red. (B) Structure of two prophage regions. Different CDS types are shown in different colours. Drug resistance genes are highlighted in blue.
Additionally, more detailed analysis of other seven CRKPs was performed using R16 as reference genome (Figure S2). We found that six CRKps (R2, R7, R12, R15, R17 and R18) might also possessed this rare plasmid (genome coverage from >77%, gene coverage >80%), and R7 had the highest homology (genome coverage 98%, gene coverage 100%). The other one CRKp strain R5 shared about 50% coverage of pR16-Hv-CRKp1.
Discussion
In this study, we describe the epidemiology of CRE in patients who presented with bacterial liver abscesses in China from 2012 to 2016, and report on the emergence of ST11-K47 and ST11-K64 Hv-CRKps. Furthermore, we also characterized the genetic basis of the drug-resistance and virulence of these strains.
Our results reveal that ST11 K. pneumoniae is the most prevalent CRKp in liver abscess cases in China. Phylogenic analysis demonstrated that CRKp and CSKp strains were clustered in different nodes, indicating that they evolved separately in distinct clonal groups. Drug resistance gene analysis showed that CRKp strains contained almost all classes of resistance genes and exhibited multidrug resistance. The emergence of such strains will lead to failure in antibacterial drug therapy and subsequent prolongation of the course of illness, resulting in increased difficulties and challenges for the treatment of infections. Earlier studies have shown that multidrug-resistant (MDR) K. pneumoniae strains have lower virulence, whereas HvKps are sensitive to most antibiotics [Citation9]. Moreover, hypervirulent and MDR K. pneumoniae strains have evolved separately in different clonal groups [Citation23]. However, our virulence assay results showed that CRE isolates, R16, R7, R17, R2, R12, R18 and R5, not only had higher virulence levels, but also contained almost all types of antibiotic resistance genes (listed in ). Unlike the Hv-CSKps that belonged to serotype K1, K2 or K57, these Hv-CRKps are K47 and K64, which has rarely been reported before. As previously reported, ST11 CRKps have been detected widely and ST11 Hv-CRKps have also been described. However, ST11 Hv-CRKps with capsular serotype K47 and K64 have rarely been reported. Until now, only two ST11-K47 Hv-CRKp have been reported and no ST11-K64 ones. All the five K47 CRKp strains showed high virulence, thus we inferred that K47 might also be a serotype related to high virulence. The emergence of ST-K47 and ST-K64 Hv-CRKps will undoubtedly present serious challenges in the management of patients infected with these strains due to its simultaneous possession of resistance and virulence genes.
Comparative analysis of virulence genes between CRKp and CSKp strains showed that the four CSKp ST23 strains (SR6, S1, S2, S8) harboured almost all virulence genes and thus exhibited higher virulence, further demonstrating that ST23 clonal group members are highly virulent. Moreover, we analyzed Klebsiella pneumoniae strains (n = 206) with complete genomes from NCBI. There were 24 ST11 K. pneumoniae strains and only two (strain 911021 and 721005) of them lacked carbapenems resistant genes. Additionally, we conducted a phylogenetic analysis and virulence/antimicrobial resistance gene distribution analysis of these two strains together with our eight ST11 CRKps (Figure S3). We found that our eight ST11 CRKps and these two CSKps were separated into two clusters, so we speculated that the ST11 CRKps and CSKps evolved in different directions, yet more work needed be done.
We also found out that K. pneumoniae strains possessing CPS regulator genes (rmpA or rmpA2), salmochelin (iroBCDN) and aerocin (iucABCD) siderophore gene clusters, had higher virulence levels as tested in NKA and G. mellonella infection assays. This was further confirmed by experiments involving another K. pneumoniae strain, K. pneumoniae1084, a hypermucoviscosity-negative K1 clinical strain, which has been shown to be of low virulence in liver abscess infection model [Citation24]. Our study demonstrated that strain 1084 lost CPS regulator gene rmpA and salmochelin and aerocin siderophore genes, unlike the high-virulence strain NTUH-K2044 (). This result further verifies the notion that these genes play important roles in the virulence phenotype of K. pneumoniae. Therefore, the acquisition of high virulence by ST11-K64 Hv-CRKp R5 is very likely to be related to the acquisition of these virulence genes. However, there were notable contradictions between virulence genotypes and phenotypes in all the five ST11-K47 strains (R16, R2, R17, R2, R12) studied, which have a highly virulent phenotype but fewer virulence genes. Nevertheless, the wzi gene and another two virulence genes, yersinibactin and mrk, were detected in all the Hv-CRKp strains although some studies indicate that they may not be HvKp specific [Citation22]. Moreover, the neutrophil killing assay and the animal experiment in the present study demonstrated that the virulence of these CRKps is comparable to Hv-CSKp strains and definitive HvKp strain K1088 [Citation9].
Homologous gene analysis against VFDB showed that four predicted virulence genes, R16_0237, R16_3744, R16_4062, R16_5486, were only existent in these CRKp strains compared with our seven CSKps (). Moreover, the predicted virulence gene, R16_3744, was also found in Kp1084 and the hypervirulent strain NTUH-K2044 (). R16_0237 shared 62.16% sequence identity with VFG035256 (ImpA-like protein); R16_3744 shared 63.83% sequence identity with VFG041224 (ctsH1, Hcp family T6SS protein CtsH1); R16_4062 shared 61.54% sequence identity with VFG044365 (fct, ferrichrysobactin receptor); and R16_5486 shared 60.61% sequence identity with VFG042772 (cofT, colonization factor antigen III). These results imply that these four predicted genes might also play important roles in the virulence of K. pneumoniae. In particular, the gene R16_5486 is located on the rare plasmid pR16-Hv-CRKp1, which simultaneously contained the carbapenemase genes blaKPC-2, ESBL genes blaCTX-M-65, blaSHV-12 and blaTEM-1B, and an aminoglycoside resistance gene rmtB. Additionally, other Hv-CRKp strains also showed high coverage of pR16-Hv-CRKp1 except for R5 (∼50% coverage), which might be the result of genomic recombination (Figure S2). To the best of our knowledge, this is the first report on this particular plasmid. We speculate that the acquisition of this plasmid might endow these strains with multidrug resistance and high virulence capacity at the same time. In contrast, two CSKps (S1 and S2) strains had a low virulent phenotype but harboured more virulence genes. In our opinion, this phenomenon occurs for multiple reasons. First, the presence of a gene is not necessarily indicative of its importance. The expression of genes is affected by multiple factors, such as gene context, interaction between different genes, antagonistic effects, and so on [Citation25,Citation26]. Moreover, SNPs or DNA modifications might also affect gene expression and function [Citation27]. Thus, more work needs to be done in this area.
Noticeably, we found that some strains showed high virulence in NKA and G. mellonella infection model but were negative for the string test (ST11-K47: R2, R7, R12, R16, and R17; ST11-K64, R5, R18; ST1764-K64, S7; ST23-K1, S1 and S2; and ), which indicates that the string test was not sensitive enough to detect the virulence of K. pneumoniae. Currently, there is still no consensus definition for HvKp and because of this, studies so far have used various criteria (e.g. string test and/or patients’ clinical features) to define these strains. The string test, which has been used widely to identify HvKp strains, has been shown to have suboptimal identification accuracy despite its handleability, particularly in low-prevalence areas [Citation28]. Clinical criteria are also problematic as the patient's profile is determined not only by the infected strain but also the patient's immune-competence and primary diseases. Moreover, notable variable consistency between the two criteria in HvKp infection has been reported from 51% to 98% [Citation29–32]. Consequently, it is difficult to make comparison between different studies as a strain defined as HvKp in one study may not qualify in another study. In this study, we used comprehensive tests combination including string test, human neutrophil killing assay and G. mellonella model to define the virulence of HvKP and the results of the latter two were concordant with each other. Nevertheless, an international consensus definition of HvKp is still needed.
Table 4. Characterization of CRE group and comparative CSE group isolates in this study.
Study limitations include possible selection bias as the geographic distribution of isolates from participating hospitals were not representative enough, with some remote regions poorly represented.
In summary, this study characterized the emergence of rare ST11-K47 and ST11-K64 Hv-CRKps, and clarified the genetic basis of drug-resistance and virulence phenotypes in CRE/CSE strains, which will provide important insights into the control and treatment of infections involving CRE or even Hv-CRE. More work is needed to understand the detailed pathogenic mechanisms and transmission dynamics of CRE/Hv-CRE strains.
Contributors
QWY, XMJ, MLZ, WHY, HZ, and YCX conceived and designed the experiments, performed the experiments, analyzed the data, and wrote the paper. Other authors provided the isolates used and approved the final version of the manuscript.
Supplemental Material
Download ()Acknowledgments
This work was supported by the National Key Research and Development Program of China (2018YFC1200100, 2018YFC1200105), Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (grant number 2016-I2M-3–014), Graduate Innovation Fund of Peking Union Medical College (grant number 2017–1002-1–21). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We are grateful to all the investigators of this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rotova V, Papagiannitsis CC, Skalova A, et al. Comparison of imipenem and meropenem antibiotics for the MALDI-TOF MS detection of carbapenemase activity. J Microbiol Methods. 2017 Jun;137:30–33. doi: 10.1016/j.mimet.2017.04.003
- Lasserre C, De Saint Martin L, Cuzon G, et al. Efficient detection of carbapenemase activity in enterobacteriaceae by matrix-assisted laser desorption ionization-time of flight mass spectrometry in less than 30 minutes. J Clin Microbiol. 2015 Jul;53(7):2163–2171. doi: 10.1128/JCM.03467-14
- Grundmann H, Glasner C, Albiger B, et al. Occurrence of carbapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing enterobacteriaceae (EuSCAPE): a prospective, multinational study. Lancet Infect Dis. 2017 Feb;17(2):153–163. doi: 10.1016/S1473-3099(16)30257-2
- Zhang R, Liu L, Zhou H, et al. Nationwide surveillance of clinical carbapenem-resistant enterobacteriaceae (CRE) strains in China. Ebiomedicine. 2017 May;19:98–106. doi: 10.1016/j.ebiom.2017.04.032
- Liu YC, Cheng DL, Lin CL. Klebsiella pneumoniae liver abscess associated with septic endophthalmitis. Arch Intern Med. 1986 Oct;146(10):1913–1916. doi: 10.1001/archinte.1986.00360220057011
- Siu LK, Yeh KM, Lin JC, et al. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012 Nov;12(11):881–887. doi: 10.1016/S1473-3099(12)70205-0
- Chang FY, Chou MY. Comparison of pyogenic liver abscesses caused by Klebsiella pneumoniae and non-K. pneumoniae pathogens. J Formos Med Assoc. 1995 May;94(5):232–237.
- Li W, Sun G, Yu Y, et al. Increasing occurrence of antimicrobial-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in China. Clin Infect Dis. 2014 Jan;58(2):225–232. doi: 10.1093/cid/cit675
- Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018 Jan;18(1):37–46. doi: 10.1016/S1473-3099(17)30489-9
- Yao H, Qin S, Chen S, et al. Emergence of carbapenem-resistant hypervirulent Klebsiella pneumoniae. Lancet Infect Dis. 2018 Jan;18(1):25. doi: 10.1016/S1473-3099(17)30628-X
- CLSI. Performance standards for antimicrobial susceptibility testing; twenty-eight informational supplement. CLSI document M100-S28. Clinical and Laboratory Standards Institute: Wayne, PA. 2018.
- Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013 Feb 15;4(2):107–118. doi: 10.4161/viru.22718
- Deleo FR, Chen L, Porcella SF, et al. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc Natl Acad Sci U S A. 2014 Apr 1;111(13):4988–4993. doi: 10.1073/pnas.1321364111
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012 May;19(5):455–477. doi: 10.1089/cmb.2012.0021
- Inouye M, Dashnow H, Raven L-A, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014 November 20;6(11):90. doi: 10.1186/s13073-014-0090-6
- Wyres KL, Wick RR, Gorrie C, et al. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb Genom. 2016 Dec;2(12):e000102.
- Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016 Jul;33(7):1870–1874. doi: 10.1093/molbev/msw054
- Chen YT, Lin JC, Fung CP, et al. KPC-2-encoding plasmids from Escherichia coli and Klebsiella pneumoniae in Taiwan. J Antimicrob Chemother. 2014 Mar;69(3):628–631. doi: 10.1093/jac/dkt409
- Zhou Y, Liang Y, Lynch KH, et al. PHAST: a fast phage search tool. Nucleic Acids Res. 2011 Jul;39 (Web Server issue):W347–W352. doi: 10.1093/nar/gkr485
- Xiong Y, Wang P, Lan R, et al. A novel Escherichia coli O157:H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS One. 2012;7(4):e36144. doi: 10.1371/journal.pone.0036144
- Rotem D, Schuldiner S. Emre, a multidrug transporter from Escherichia coli, transports monovalent and divalent substrates with the same stoichiometry. J Biol Chem. 2004 Nov 19;279(47):48787–48793. doi: 10.1074/jbc.M408187200
- Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019 Jun 19;32(3):e00001–19. doi: 10.1128/CMR.00001-19
- Hennequin C, Robi F. Correlation between antimicrobial resistance and virulence in Klebsiella pneumoniae. Eur J Clin Microbiol Infect Dis. 2016;35(3):333–341. doi: 10.1007/s10096-015-2559-7
- Lin AC, Liao TL, Lin YC, et al. Complete genome sequence of Klebsiella pneumoniae 1084, a hypermucoviscosity-negative K1 clinical strain. J Bacteriol. 2012 Nov;194(22):6316. doi: 10.1128/JB.01548-12
- Winther-Larsen HC, Josefsen KD, Brautaset T, et al. Parameters affecting gene expression from the Pm promoter in gram-negative bacteria. Metab Eng. 2000 Apr;2(2):79–91. doi: 10.1006/mben.1999.0142
- Geissmann T, Possedko M, Huntzinger E, et al. Regulatory RNAs as mediators of virulence gene expression in bacteria. Handb Exp Pharmacol. 2006;173:9–43. doi: 10.1007/3-540-27262-3_2
- Shell SS, Prestwich EG, Baek SH, et al. DNA methylation impacts gene expression and ensures hypoxic survival of mycobacterium tuberculosis. PLoS Pathog. 2013;9(7):e1003419. doi: 10.1371/journal.ppat.1003419
- Russo TA, Olson R, Fang CT, et al. Identification of biomarkers for differentiation of hypervirulent Klebsiella pneumoniae from classical K. pneumoniae. J Clin Microbiol. 2018 Sep;56(9):e00776–18. doi: 10.1128/JCM.00776-18
- Fang CT, Chuang YP, Shun CT, et al. A novel virulence gene in Klebsiella pneumoniae strains causing primary liver abscess and septic metastatic complications. J Exp Med. 2004 Mar 1;199(5):697–705. doi: 10.1084/jem.20030857
- Lee CH, Liu JW, Su LH, et al. Hypermucoviscosity associated with Klebsiella pneumoniae-mediated invasive syndrome: a prospective cross-sectional study in Taiwan. Int J Infect Dis. 2010 Aug;14(8):e688–e692. doi: 10.1016/j.ijid.2010.01.007
- Lin YC, Lu MC, Tang HL, et al. Assessment of hypermucoviscosity as a virulence factor for experimental Klebsiella pneumoniae infections: comparative virulence analysis with hypermucoviscosity-negative strain. BMC Microbiol. 2011 Mar 8;11:50. doi: 10.1186/1471-2180-11-50
- Yu WL, Ko WC, Cheng KC, et al. Comparison of prevalence of virulence factors for Klebsiella pneumoniae liver abscesses between isolates with capsular K1/K2 and non-K1/K2 serotypes. Diagn Microbiol Infect Dis. 2008 Sep;62(1):1–6. doi: 10.1016/j.diagmicrobio.2008.04.007