
ABSTRACT
With increasing frequency, humans are facing outbreaks of emerging infectious diseases (EIDs) with the potential to cause significant morbidity and mortality. In the most extreme instances, such outbreaks can become pandemics, as we are now witnessing with COVID-19. According to the World Health Organization, this new disease, caused by the novel coronavirus SARS-CoV-2, has already infected more than 10 million people worldwide and led to 499,913 deaths as of 29 June, 2020. How high these numbers will eventually go depends on many factors, including policies on travel and movement, availability of medical support, and, because there is no vaccine or highly effective treatment, the pace of biomedical research. Other than an approved antiviral drug that can be repurposed, monoclonal antibodies (mAbs) hold the most promise for providing a stopgap measure to lessen the impact of an outbreak while vaccines are in development. Technical advances in mAb identification, combined with the flexibility and clinical experience of mAbs in general, make them ideal candidates for rapid deployment. Furthermore, the development of mAb cocktails can provide a faster route to developing a robust medical intervention than searching for a single, outstanding mAb. In addition, mAbs are well-suited for integration into platform technologies for delivery, in which minimal components need to be changed in order to be redirected against a novel pathogen. In particular, utilizing the manufacturing and logistical benefits of DNA-based platform technologies in order to deliver one or more antiviral mAbs has the potential to revolutionize EID responses.
Introduction
Unmet needs of emerging infectious disease outbreaks
Fuelled by multiple factors, including the growing urbanization of society, climate change, and shifting agriculture and forestry practices, and fanned by expanded global travel and trade, there is no doubt that we are in a period of increasing viral outbreaks [Citation1,Citation2]. In just the past two decades, novel viral outbreaks have caused significant damage with wide-reaching ramifications. From the severe acute respiratory syndrome (SARS) outbreak in 2003, the H1N1 influenza A virus (IAV) pandemic in 2009, the Middle East respiratory syndrome (MERS) outbreak in 2012, the recurring Ebola virus outbreaks in 2014–2016, to the expansion of chikungunya virus and the rapid spreading of Zika virus infections in 2015, viral diseases were already a widely acknowledged threat even before the current pandemic. When faced with a virulent, rapidly spreading EID, and as the current COVID-19 situation attests, there is a limited window of opportunity for successfully halting the spread of an outbreak with pandemic potential. In the case of new outbreaks of known viruses, particularly those for which there are diagnostic tools as well as vaccines or antiviral drugs, an effective response entails a combination of epidemiological detective work as well as treating and preventing new infections. By testing for and tracing the pattern of infections, health care workers can identify and isolate people as needed, and when available, prevent new infections through vaccination. Antiviral drugs have the potential to lessen the burden on the health care system by reducing disease severity, including in the health care workers themselves, as well as reduce transmission by lowering the viral load in infected individuals. Indeed, the use of highly active antiretroviral therapy to significantly reduce HIV-1 plasma viremia essentially abolishes sexual transmission of the virus [Citation3].
In the case of an outbreak of a pathogenic virus for which there is no available vaccine or drug, the initial responses are limited to supportive medical care for patients and physical barriers, such as quarantines and the use of face masks for respiratory diseases, to reduce transmission. These techniques vary in efficacy, however, and in the case of a new pathogen for which critical details like the transmission route and incubation and infectious periods are not yet known, they are hampered by a lack of information. The exact duration of the window of opportunity for preventing an outbreak from becoming a pandemic varies considerably between different viruses and host populations, due primarily to the R0, or basic reproductive number of the virus, and the rate of transmission [Citation4]. What is ultimately needed is a vaccine or antiviral drug to reliably stop the spread of infection, and if one does not yet exist, a new viral countermeasure must be developed in a race against time ().
Figure 1. Overlay of cumulative confirmed global COVID-19 cases as of June 19, 2020 as reported by the World Health Organization, with the projected accelerated timelines for developing various antiviral countermeasures. These clinical evaluation timelines have been rapidly accelerated to meet the constraints of COVID-19. Repurposing existing drugs would be the fastest way to treat COVID-19, assuming efficacy. Vaccines are expected to be the slowest as there is a high burden to demonstrate safety and efficacy. The timelines represent a projected average for each phase of clinical development.
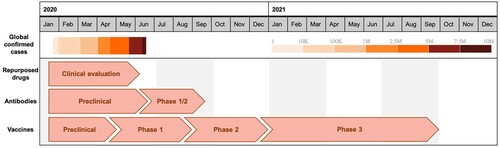
Recent outbreaks have revealed that our ability to develop such countermeasures is too slow [Citation5]. In response to an Ebola virus outbreak in West Africa that began in 2014, it took until the end of 2019 for a vaccine (Ervebo) to be granted approval by the U.S. Food and Drug Administration (FDA) for the prevention of Ebola virus disease (EVD). This was despite significant effort and investment by both the public and private sectors [Citation6]. Because of the existing infrastructure for influenza vaccine development, vaccines against the 2009 H1N1 pandemic IAV managed to gain approval more quickly, by September of that same year, but manufacturing issues prevented widespread availability of a vaccine until after the peak of the outbreak [Citation7]. The emergence of SARS-CoV in 2002 also prompted efforts to develop a vaccine, but none have yet been successful.
An efficacious vaccine is the gold standard for public health, with the greatest ability to reduce the number of infections, thereby limiting the scope of societal and economic damage. Unfortunately, however, vaccines typically take a long time to develop [Citation8]. Vaccines against infectious diseases can be largely grouped into those that deliver just a specific target antigen (a viral glycoprotein, for example) or the entire pathogen in an inert or weakened form (such as heat-inactivated or live-attenuated virus). Once the preliminary safety and efficacy studies are completed in animals, extensive clinical testing is necessary in order to establish that the vaccine is truly both safe and effective in humans. Following regulatory approval, large scale manufacturing and quality control are undertaken to generate the product for distribution. In general, this entire process takes from a few to several years (or more), a timeframe unlikely to be helpful in the case of an acute, rapidly spreading EID. In recognition of this, organizations such as the Coalition for Epidemic Preparedness Innovation (CEPI) aim to reduce that time; for CEPI that entails developing accelerated programmes capable of moving from identification of a novel pathogen to the beginning of clinical trials in just 16 weeks [Citation9].
The development timeline for novel antiviral drugs is also typically far longer than what would be useful for containing a rapidly growing outbreak. The entire process, from target identification and validation, screening and lead identification, and preclinical animal studies, to scaled-up manufacturing, clinical trials, and regulatory approval, is estimated to take an average of 10–15 years [Citation10]. The most promising prospect for antiviral drugs in the case of an EID is the possibility that a drug which has already been approved for another use can be repurposed. In that case, only a clinical efficacy study would be needed (and possibly scaled-up manufacturing). Indeed, such studies are currently underway to assess the efficacy of a number of existing antivirals for treating COVID-19, including Avigan (favipiravir), a drug used for IAV treatment, and Kaletra (lopinavir/ritonavir), a drug combination that inhibits HIV-1 [Citation11–13]. Remdesivir, which had already been through clinical safety studies as part of an effort to test its potential as a treatment for EVD, showed some benefit in shortening the time to recovery from severe COVID-19 disease and has recently received an emergency use authorization from the FDA [Citation14].
Antibodies as drugs for fighting infectious disease
Monoclonal antibodies (mAbs) represent a unique class of biologic antiviral drugs. The vast majority of FDA-approved mAbs have been developed to treat one or more of a wide range of cancers and inflammatory diseases. A few, however, have been approved for use in infectious disease settings, such as palivizumab for respiratory syncytial virus and ibalizumab-uiyk for the treatment of multidrug-resistant HIV-1 infection [Citation15]. The potential of mAb use for protection against viral diseases has increasingly been raised, partly due to the recent surge in clinically approved mAbs in other disease areas as well as advancements in mAb technologies in general [Citation15,Citation16]. In acute settings, such as in response to an EID or bioterrorism, where the speed of conferring protection is of particular value, the potential for mAb use has been widely acknowledged [Citation15,Citation17]. In contrast to vaccines, in which protection takes several days to a few weeks to fully develop, mAbs have the ability to provide near instantaneous protection following administration [Citation17,Citation18].
Broadly speaking, the process of developing a mAb as an antiviral drug is much like that of a small molecule, but with some notable exceptions. First, because mAbs are naturally produced by B cells within the body, primarily for defense against pathogens, much is known about their general physical and chemical properties in vivo. Furthermore, new mAbs can be made using a standard mAb backbone, including ones that have been previously characterized in clinical studies. Many recent technological advances in single-cell sequencing have resulted in rapid identification of hundreds to thousands of antigen-specific mAbs against a number of targets, and optimization and down selection of antiviral mAbs identified in this way was recently reported to be performed in as little as 10 days [Citation19]. Another particularly useful characteristic of using mAbs as drugs is the ability to imbue them with specific functions by modulating the amino acid sequence of the Fc region. For example, the ability to bind (or not) and engage Fc receptors on various immune cells can be rationally designed into the mAb Fc domain [Citation20]. This is of particular importance in infectious disease settings that are susceptible to Fc receptor-mediated antibody dependent enhancement (ADE) of infection. Indeed, ADE has been well-documented for dengue viruses and Zika virus among others, and has been proposed as a possible concern for the coronavirus diseases SARS and MERS as well [Citation21–23].
Despite these many advantages of mAbs as antiviral drugs, they are very expensive to manufacture [Citation24]. The process involves expression in mammalian cells, whose growth must be scaled up to large bioreactors, followed by extensive purification and finishing steps to ensure the product is ready for clinical use [Citation25]. This typically takes anywhere from 9 to 18 months, although there are efforts to make this timeline shorter, particularly with respect to mAb production for EID responses. Technological advances in combination with process changes, such as running some activities in parallel that would normally be conducting sequentially, may have the potential to bring the time from lead mAb identification to production of material for clinical evaluation down to 5–6 months [Citation26]. Regardless, the high costs associated with mAb manufacturing in part underlie the extremely high market price for these molecules; an analysis of all 107 FDA-approved mAbs through 2016 revealed a mean and median annual price of $96,731 and $58,968, respectively [Citation27]. Though antiviral mAb prevention or treatment regimens may require less mAb protein than a typical year’s worth of mAb used for chronic diseases, the cost is seen as a major barrier to the more widespread use of mAbs for infectious disease [Citation17]. However, the use of mAbs as therapeutics in acute disease settings, such as in cases of severe COVID-19, remains an important driver for their continued development.
Accelerating the response with nucleic acid-based delivery
Despite the traditionally long timelines for new vaccine and antiviral drug development, these established strategies for preventing and treating viral infections have clearly had many successes, not the least of which is the global eradication of smallpox and massive reductions in the number of other viral diseases like polio and measles through vaccination [Citation28]. Similarly, the development of a vast array of antiviral drugs for treating HIV-1 infections resulted in an estimated gain of 14.4 million life-years by 2009 [Citation29]. None of these interventions, however, came in time to prevent these viral outbreaks from spreading throughout the globe. And so, despite these successes, the increasing threats from novel viruses have raised serious concerns about the significant gap between the current timeframe for the development of new vaccines or antiviral drugs and the timeframe that would actually be needed to prevent an EID from becoming a pandemic. A straightforward way to facilitate the development of new antiviral countermeasures would be through the establishment of a platform technology, such that only one or a few components would need to be changed to redirect the specificity of a proven antiviral strategy [Citation30].
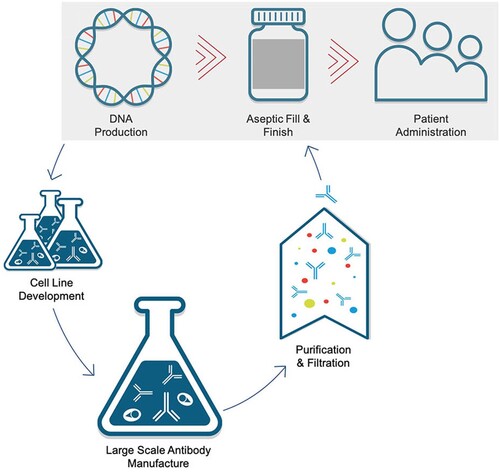
One of the more prescient efforts to close this gap, and another acknowledgement that a platform technology is the best strategy for fast responses to an EID, was begun by an agency within the United States Department of Defense, the Defense Advanced Research Projects Agency (DARPA). In early 2017, DARPA released an announcement for funding a programme called the Pandemic Prevention Platform, or P3, Program. This announcement detailed funding to support the development of a platform technology capable of moving from the identification of a novel viral pathogen, to the manufacture and readiness to deliver 20,000 doses of a countermeasure within 60 days. Aside from the astounding strategic and technological innovations required for accomplishing in two months what would normally take years, there were two key features of the programme that focused the scope of the work they wanted to fund. First, they were predominantly looking for proposals that would identify and optimize mAbs capable of binding to a virus in a way that prevents infection (so-called neutralizing antibodies or NAbs) for use as the viral countermeasure. These special mAbs typically work by binding to the viral protein exposed on the outer surface of the virion that is used by the virus to gain entry to a cell. Through either direct inhibition of cellular attachment or indirectly interfering with the conformation of these viral proteins, at sufficient concentrations such NAbs are able to render a viral particle largely inert [Citation31]. And although historically the delivery of mAbs for infectious disease prevention and therapy has been a much smaller area of research than vaccines and antiviral drug discovery, the aforementioned dramatic improvements in technologies for mAb identification and optimization have rapidly accelerated the field such that it has the potential to meet the short timeline required for containing an EID. The second key feature of the P3 programme stipulated that in order to manufacture countermeasures within the requisite timeframe, the delivered product would need to be a nucleic acid encoding the NAb, rather than the purified NAb protein itself. By delivering the NAb genes directly to people, all of the protein production and purification steps can be eliminated from the manufacturing process, essentially enabling the body to serve as the bioreactor ().
Figure 2. DNA-based medical countermeasures bypass multiple manufacturing steps required with traditional antibody therapeutics. Generating antibody drug product requires cell line development followed by large scale production in bioreactors, and multistep purification and filtration in advance of vialing the product. DNA-based antibody delivery (highlighted in the gray box) circumvents many of these costly and timeconsuming steps.
Main text
In vivo expressed biologics: the body as a bioreactor
Nucleic acid-based NAb delivery can be achieved using one of three main technologies: Adeno-associated virus (AAV) vectors, lipid nanoparticle (LNP)-formulated mRNA, or in vivo transfection of naked DNA facilitated by electroporation (DNA/EP). There are variations of each of these three types of technologies, but they are broadly differentiated by manufacturing requirements, administration requirements, levels of expressed gene products, and duration of expression. Other issues regarding safety profiles, immunogenicity, and redosing are also important variables to consider when comparing and contrasting these methods. Regardless of the delivery system, the goal of all nucleic acid-based mAb platforms is expression and production of the mAb by the target cells (e.g. skeletal muscle cells or hepatocytes, depending on the route of administration), resulting in secretion of mAbs and eventual uptake by the vascular system [Citation32].
The majority of work in the AAV field is focused on gene therapy and protein replacement applications. In order to generate an AAV-based vector, an expression cassette containing the gene of interest is inserted into the AAV genome, replacing the viral genes but retaining the genomic structural elements required for encapsulation by the virion as well as stability and persistence within the cell. This modified DNA genome is then packaged into AAV in cells expressing the viral genes necessary for producing viral particles, purified from the culture system, and delivered typically either by intravenous (IV) infusion or intramuscular (IM) injection [Citation33]. The first AAV vectors for gene delivery were developed in the mid-1980s and the first clinical use was in 1995 for the treatment of cystic fibrosis. The first regulatory approval didn’t come until 2012, however, when the European Medicines Agency gave the go-ahead for Glybera, an AAV-based treatment of hereditary lipoprotein lipase deficiency. The first FDA approval for AAV came in 2017 for Luxturna, a treatment for a specific type of inherited retinal disease. Only one other AAV therapy (Zolgensma, for spinal muscular atrophy) has received FDA approval since, though there are hundreds currently in clinical trials [Citation33]. Despite the large interest in AAV-based therapies, the high costs associated with AAV manufacturing are in part reflected in the prices of these drugs and negatively affect their potential for more widespread clinical use [Citation33]. Indeed, Luxturna was priced at $425,000 per eye when it first came on the market in 2017, and Zolgensma set a world record with its $2.1 million price tag in 2019.
Only two clinical trials have evaluated AAV-mediated delivery of NAbs. The first, a phase 1 study to evaluate the delivery of the anti-HIV-1 NAb, PG9, began in 2014, but unfortunately resulted in undetectable levels of PG9 in patient serum [Citation34]. The second, begun in 2018, was designed to evaluate another anti-HIV-1 NAb, VRC07 (www.clinicaltrials.gov Identifier: NCT03374202). In contrast to the PG9 study, two out of three participants receiving the highest AAV dose maintained ∼0.25–1.25 µg/mL serum concentrations of VRC07 for at least 40 weeks post administration [Citation35]. Small animal models of AAV-mediated delivery are capable of achieving remarkable serum concentrations (>100 µg/mL) of circulating NAb, sometimes for many months or even years, but have struggled with the frequent appearance of NAb-depleting immune responses [Citation36], likely a significant factor in the PG9 trial [Citation34]. However, important proof-of-concept was established in a monkey, in which the expression of two potent anti-HIV NAbs (3BNC117 and 10-1074) durably suppressed the viral load of a simian-tropic HIV-1 (SHIV)-infected rhesus macaque following AAV-mediated delivery, though this study also faced confounding results due to issues with immunogenicity [Citation37]. Although the results thus far from the VRC07 clinical trial are promising, it remains to be seen whether AAV technology can robustly scale to large animals and humans.
The majority of mRNA delivery strategies use a type of lipid-based nanoparticle in order to stabilize and protect the mRNA molecules, as well as facilitate cell entry [Citation38]. Though usually delivered systemically by IV infusion, some mRNA delivery technologies are exploring direct, tissue-specific delivery to the heart, lungs, or tumors, for example. Thus far, most of the work on mRNA delivery has been focused on vaccination, in which the delivered RNA encodes an antigen designed to elicit a specific immune response. More recently, however, interest in using mRNA for protein therapy has increased and is currently being investigated in several clinical trials. The first clinical trial for evaluating an mRNA-based delivery system of an antiviral NAb, however, was initiated just last year by Moderna (www.clinicaltrials.gov Identifier: NCT03829384), for an antibody directed against chikungunya virus.
In contrast to the long-lived expression often seen with AAV-based gene transfer, mRNA delivery has been largely characterized by much shorter mAb persistence, typically not longer than a few weeks [Citation39]. In many of these studies, however, the durability of the serum NAb titer was confounded by the immunogenicity of non-species matched elements of the NAbs. LNP-mRNA has successfully been used to deliver NAbs against HIV-1, influenza B virus, or rabies virus in mice, resulting in peak titers of ∼10–150 µg/mL at ∼1 day post IV administration[Citation40,Citation41]. In cynomolgus monkeys, LNP-mRNA-mediated delivery of a NAb against IAV reached a peak serum concentration of ∼4 µg/mL, also at 1 day post IV administration [Citation42]. Many of the recent advances in LNP-RNA technology lie in the development of new LNP formulations that enhance their efficacy and safety, however it is largely these formulations that make the manufacture and quality control of the particles challenging [Citation39].
Clinically, DNA/EP technology has predominantly been used for DNA-based vaccines. Similar to mRNA vaccines, a gene encoding an antigen is delivered with the hope of eliciting a specific immune response to the expressed gene product. However, in the case of DNA/EP, the antigen coding sequence is inserted into a plasmid DNA (pDNA) expression vector and injected via either the IM or intradermal route. The subsequent electrical pulses facilitate the uptake of the pDNA into cells as well as enhance immune responses, thereby greatly improving the vaccine efficacy as compared to injection of pDNA alone [Citation43]. The use of DNA/EP for protein therapy or NAb delivery is largely still a preclinical field, although the intratumoral delivery of pDNA-encoded interleukin-12 by electroporation has shown promise in the clinic, most notably in treating melanoma [Citation44]. The first clinical trial evaluating DNA/EP-mediated delivery of an antibody is currently underway in a study evaluating an anti-Zika virus NAb (www.clinicaltrials.gov Identifier: NCT03831503).
Although injection of pDNA alone can result in some transgene expression, the levels are generally weak, and for systemic mAb delivery, result in very low serum protein concentrations. In the search for improved transfection efficiency, and because EP was already well established as a method of significantly improving in vitro transfection, EP was an obvious candidate for a method of improving in vivo transfection. Indeed, even in the nascent days of DNA/EP technology development, the addition of electrical pulses resulted in more than a 100-fold increase in peak serum protein levels as compared to pDNA injection alone [Citation45]. As early as the late 1990s, in vivo electroporation was evaluated for gene transfer studies of interleukin-5, fibroblast growth factor 1, and erythropoietin in mouse studies [Citation45–47]. Skeletal muscle was the target tissue of choice for each of these studies, as it was recognized that “the use of highly vascularized muscle as an endocrine organ for the systemic secretion of therapeutic proteins” held much promise [Citation46]. It was not much longer before the demonstration that this same process could be applied equally to the delivery of antibodies, either as therapeutic agents or for the prevention of infectious disease () [Citation48,Citation49].
Table 1. DNA/EP-mediated delivery of antiviral mAbs for infectious disease prophylaxis.
Though some of the mechanistic details of the overall process remain incompletely understood, the general principle of in vivo EP is similar to that for in vitro EP, in that the application of an electrical field of sufficient strength is able to facilitate the uptake of extracellular pDNA by target cells [Citation50]. The electrodes and electrical conditions used in vitro, however, do not readily translate to an in vivo setting where the extracellular environment, structural elements of three-dimensional tissues, and immune system surveillance are vastly different. Thus, much effort has been put into evaluating different electrodes and electrical conditions for maximizing transgene expression, while minimizing muscle damage [Citation51–56]. For protein therapy applications, where the demands for maximizing expression levels and protein production are typically higher than for vaccines, the pDNA is almost always delivered via IM injection, rather than intradermally. Following the injection of the pDNA, electrical pulses are delivered either with plate electrodes, which remain external, or needle electrodes, which are inserted into the muscle adjacent to the pDNA injection site. The key electrical parameters are the voltage, pulse duration, and pulse number. Most current DNA/EP studies deliver 1–4 pulses for 10–400 ms at 100–250 V/cm. Some groups have also experimented with higher voltage pulses, in the 500–1500 V/cm range, but these pulses are typically much shorter, in the 5–100 µs range. Regardless of the exact pulsing protocol, the overall procedure is quite fast and the pDNA injection and electrical pulse delivery can be completed in a matter of a minute or less.
Once the general DNA/EP procedure was established, a number of proof-of-concept animal studies were carried out in order to demonstrate that proteins produced by this delivery method were indeed functional. The first of these demonstrated that the delivery of erythropoietin by DNA/EP led to an increase in hematocrit and a subsequent study revealed the benefit of cardiotrophin-1 delivery for progressive motor neuron disease [Citation47,Citation57]. It was nearly a decade later, however, before the first demonstration that DNA/EP-mediated delivery of a NAb could protect mice from a lethal IAV challenge [Citation58]. In another important model of DNA/EP-mediated infectious disease prophylaxis, the delivery of an anti-dengue virus NAb bearing an Fc mutation that abrogates FcγR binding was able to protect mice from viral challenge, even in the presence of a known ADE-inducing, non-neutralizing anti-dengue virus mAb [Citation59]. A number of subsequent studies demonstrated the broad potential for DNA/EP-mediated delivery of NAbs for the prevention of infectious diseases, including not only IAV, but influenza B virus, chikungunya virus, Ebola virus, and Zika virus [Citation60–65.] Importantly, the Zika virus studies also established proof-of-concept for protection in non-human primates (NHPs) [Citation64,Citation65].
An important advantage revealed by some of these studies of infectious disease prophylaxis, but with relevance to other disease indications that may benefit from mAb combination therapy (such as trastuzumab with pertuzumab for HER2+ breast cancer), is the relative ease of delivering multiple mAbs by DNA/EP. For some infectious diseases, such as HIV-1, a number of broadly reactive and very potent NAbs have been identified, so-called broadly neutralizing antibodies (bNAbs), which have the potential to single-handedly prevent or treat infection by a wide range of virus strains () [Citation66,Citation67]. Even these remarkable molecules, however, work most effectively in combination, particularly as a means to reducing the emergence of resistant viruses. Combinations of either three or five bNAbs were able to potently suppress HIV-1 replication in humanized mice, and in contrast to bNAb monotherapy, greatly reduced the emergence of viral resistance through the acquisition of escape mutations [Citation68]. With recent advancements in antiviral mAb identification and characterization, there is now the opportunity to rapidly identify multiple mAbs of varying specificity and potency against a given viral pathogen. Subsequent combinatorial analysis of different pools of mAbs can then be used to rapidly identify mAb groups that display enhanced overall breadth and potency. For example, in depth analysis of different combinations of bNAbs that target distinct sites on the HIV-1 envelope glycoprotein revealed combinations that yield substantially improved neutralization breadth in vitro [Citation69]. These data can then be used to inform in vivo studies, where the optimal combination can be delivered to achieve the most significant antiviral effect. Indeed, as novel NAbs are discovered, they can be plugged into such systems to rapidly identify the current best NAb combinations for use against a given virus.
Figure 3. Schematic illustrating breadth and potency characteristics of antibodies overlaid with difficulty of eliciting specific properties.
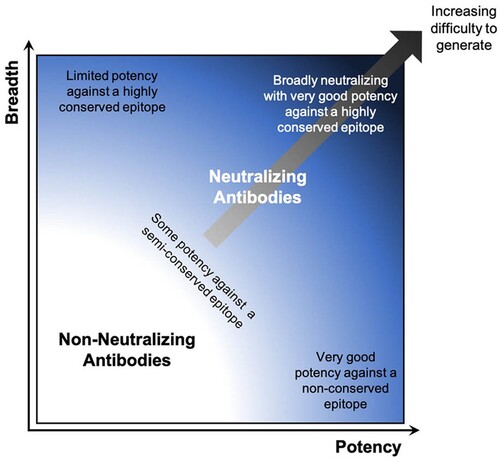
Because combinations of NAbs have the ability to significantly enhance the overall efficacy of mAb therapy, it opens the door to mAb use against other viruses for which few, if any, mAbs have been identified that are potent and broadly protective enough to be used on their own. In these cases, pooling two or more standard NAbs, which arise more frequently than bNAbs and are therefore easier to isolate (), is a strategy that can achieve the same effect. In fact, such combinations better reflect the natural polyclonal Ab responses generated by vaccination or infection. A number of animal studies have demonstrated the effective delivery of multiple mAbs by DNA/EP, including pools of anti-viral NAbs against IAV, HIV-1, and Ebola virus [Citation62,Citation63,Citation70]. Importantly, the delivery of a combination of three NAbs targeting distinct sites on the IAV hemagglutinin protein provided complete protection from either H1N1 or H3N2 IAV-associated mortality, in contrast to the moderate protection conferred by an equivalent amount of the individual NAbs administered as single agents [Citation62]. Indeed, this is the basis for the triple antibody cocktails ZMapp and REGN-EB3 recently investigated in clinical trials for the treatment of Ebola virus disease [Citation71]. Additionally, incorporating NAbs with activity against distinct viruses, such as those targeting both SARS-CoV-1 and -2, raises the possibility of assembling NAb pools that can not only provide protection against a known EID, but may also serve as a ready countermeasure against an as yet unknown member of the same virus family [Citation72,Citation73].
The potential benefits of multiple mAb delivery extend beyond just combining antiviral NAbs. Indeed, mAbs with therapeutic benefits outside of direct virus neutralization could enable a multi-mechanistic approach to fighting particular viral infections. For example, a surface glycoprotein-binding NAb paired with an immunomodulatory mAb could act to both lower viral load and boost the immune response. Alternatively, for viral infections marked by pathologies associated with excessive immune activation, NAbs could be combined with agents to reduce inflammation. This latter strategy is particularly relevant to the current COVID-19 pandemic, where elevated immune responses are associated with severe disease in some patients [Citation74]. Indeed, tocilizumab, a mAb antagonist of the IL-6 receptor, and anakinra, an interleukin-1 receptor antagonist, are being evaluated for off-label use in COVID-19 patients [Citation75,Citation76].
Given the high cost of mAb manufacturing, the price of giving multiple NAbs by standard protein administration methods is likely to be prohibitive in many cases. Likewise, the expense and difficulty of both AAV and LNP-mRNA manufacturing limits the feasibility of delivering multiple NAbs with these methods. The relatively lower cost of pDNA production, however, makes this highly attractive strategy of administering NAb cocktails for infectious disease a much more realistic possibility, particularly when the goal is to administer them to large numbers of people as a prophylactic.
Remaining questions for DNA-based antiviral mAb delivery
Although there is little clinical data surrounding DNA/EP for the purpose of mAb delivery, numerous vaccine studies using DNA/EP devices have demonstrated overall positive safety and tolerability profiles for this technology in general [Citation75,Citation77]. The previous clinical experiences of these devices are no doubt part of the reason the sole clinical trial for the DNA/EP-mediated mAb gene transfer of a therapeutic mAb is being conducted with a device designed for DNA vaccination [Citation31]. This phase 1 trial, sponsored by the University of Pennsylvania and Inovio Pharmaceuticals, began in early 2019 in order to investigate the safety, tolerability, and serum antibody concentrations of an anti-Zika virus NAb delivered by DNA/EP. While there is hope that the data from this first trial will demonstrate sufficient serum mAb concentrations in the study participants, currently it remains to be seen whether the technology is capable of that yet.
One of the most significant hurdles between the preclinical and clinical stage of DNA/EP use for mAb delivery is scaling from small animal models to humans. The first human clinical trial designed to evaluate DNA/EP for mAb delivery was unique in that the antibody was designed to act as a vaccine, rather than as a prophylactic or therapeutic agent (www.clinicaltrials.gov Identifier: NCT01138410), and so systemic levels of the delivered mAb were not an important outcome. Therapeutically relevant serum mAb concentrations are readily achieved by DNA/EP in murine proof-of-concept models where a relatively large volume of muscle tissue per body surface area can easily be targeted. Additionally, the blood volume in which the mAb gets diluted as it enters systemic circulation is relatively small in a mouse, around 2.5 mL. In contrast, the average blood volume of an adult human is ∼6000 mL, a ∼2400-fold increase that greatly exceeds the relatively small increase in allowable IM injection volume: 0.05 mL for mice and ∼1 mL for humans (depending on the muscle), a mere ∼20-fold increase. As a result, the overall process needs to be more efficient in order to achieve the same serum mAb levels in humans as in small animal studies, and thus such studies do not accurately predict the levels that are achievable in people.
There are multiple routes for improving upon the DNA/EP process, including device modifications, electrical parameter optimizations, plasmid vector engineering, as well as changes to the pDNA formulation. For example, many studies, including the current phase 1 trial, rely on the enzyme hyaluronidase, either as a pre-treatment or co-formulated with the pDNA. Hyaluronidase, which is commercially available for clinical use as Hylenex, breaks down hyaluronic acid in the extracellular matrix and is thought to facilitate the diffusion of the injected pDNA within the muscle prior to the delivery of electrical pulses.
In order to realistically interpret improvements in DNA/EP efficiency and better understand the changes that are most relevant for scaling to humans, it is often useful to evaluate large animal models as intermediates between mouse and human scales. Studies in rabbits and macaques, as well as larger animals such as sheep and pigs, have enabled a much clearer look at whether current DNA/EP devices and procedures are capable of delivering sufficient mAb levels in humans. NHPs are often viewed as the most clinically relevant animal model, and while they may be the most relevant from a safety perspective, their relevance to addressing issues of scale is somewhat limited. Studies performed in NHPs to date have been performed with macaques weighing an average of 4–10 kg, and therefore still far from humans. However, even at this size, macaques are large enough to permit the use of a human or near-human-sized EP device. Significant results have been achieved thus far in the DNA/EP-mediated delivery of mAb to NHP; most impressively, serum mAb concentrations of 6–34 µg/mL were measured in cynomolgus macaques receiving pDNA encoding the anti-HIV-1 bNAbs PGDM1400 and/or PGT121 [Citation70].
Early studies in 15–17 kg sheep with various devices were less promising, resulting in very low serum mAb levels (∼25–150 ng/mL) that peaked 1–2 weeks after delivery and quickly fell to undetectable levels by 4 weeks [Citation48,Citation78]. Both of these early sheep studies delivered murine mAbs, however, and so the persistence of serum mAb was curtailed by host immune responses. More recent studies in much larger 40–70 kg sheep were able to attain 300 ng/mL mAb in plasma, and because these studies were performed with fully ovine mAbs, the levels remained detectable for three months [Citation79]. Hyaluronidase pre-treatment boosted the mAb concentration to 1.8 µg/mL, however, immune responses directed against the mAb variable regions were observed in animals in which these high mAb levels were achieved [Citation79].
The transient mAb levels observed in some of these studies highlight a common problem in many animal studies to date; the use of non-species matched antibodies significantly impairs the ability to accurately assess the PK profile of the expressed mAbs after delivery. This is primarily due to the fact that the majority of non-species matched mAbs quickly elicit a potent immune response, largely characterized by antibodies directed against the delivered mAb. The appearance of these so-called anti-drug antibodies (ADAs), a relatively common occurrence with many therapeutic proteins, can lead to rapid protein depletion [Citation80]. This makes an extrapolation of the PK profile as it might be in a species-matched setting impossible. Such is the case when expressing a human mAb in mice in which serum mAb levels usually decline rapidly within a few weeks, whereas the delivery of a murine mAb can result in several months of readily detectable expression [Citation62].
Even in the absence of confounding factors like ADAs, the levels of different mAbs can vary significantly. Indeed, in the evaluation of two ovine mAbs differing only in their variable regions, there were ∼3-fold differences in the peak mAb levels observed [Citation79]. These results highlight the influence of the mAb sequence on both mAb expression and ADA response. Importantly, however, the sheep used in these studies were nearly an order of magnitude larger than the NHPs used to evaluate human mAbs, and were able to reach therapeutically relevant (single digit µg/mL) mAb levels, thereby demonstrating real promise for the ability of DNA/EP to achieve such levels in humans. It should be noted, however, that the DNA/EP administration was performed at a minimum of 12 sites per animal in order to reach these levels in sheep, clearly something that will need to be reduced prior to successful human use.
Closing remarks
In responding to an EID, such as COVID-19, all parts of the drug discovery and development process must be sped up. Barring the best-case scenario, in which approved, existing drugs can be repurposed, research into new viral countermeasures must begin as soon as the threat is discovered. Regulatory paths must also be streamlined, as the FDA has recently done by aggressively moving to increase the pace of the review and approval processes through the creation of a novel programme, the Coronavirus Treatment Acceleration Program. Technological advances have already greatly accelerated the science, from discovery through manufacturing, in part due to government initiatives like the P3 programme and organizations like CEPI. The development of platform technologies, and in particular, nucleic acid-based technologies, offer the best hope for rapid responses to novel viral outbreaks. Indeed, DARPA recently launched a new programme, Nucleic Acids On-Demand Worldwide (NOW), aimed at driving innovation in nucleic acid manufacturing technologies, to further reduce the EID response timeline. Combining the developability advantages of DNA/EP in particular as a nucleic acid-based platform technology with the power of mAbs as antiviral drugs, especially when delivered as potent cocktails, may represent the best hope of containing the next EID. It is inevitable that we will encounter more viral outbreaks with pandemic potential, what is not inevitable is that such outbreaks will actually become pandemics.
Disclosure statement
C.D.A. and R.A.L. are employees of RenBio, a company developing a DNA/EP platform technology for antibody delivery. Y.H. and D.D.H. are co-founders of RenBio.
References
- Smith KF, Goldberg M, Rosenthal S, et al. Global rise in human infectious disease outbreaks. J Roy Soc Interface. 2014;11:20140950. doi: 10.1098/rsif.2014.0950
- Findlater A, Bogoch II. Human mobility and the global spread of infectious diseases: a focus on air travel. Trends Parasitol. 2018;34:772–783. doi: 10.1016/j.pt.2018.07.004
- Eisinger RW, Dieffenbach CW, Fauci AS. HIV viral load and transmissibility of HIV infection. Jama. 2019;321:451–452. doi: 10.1001/jama.2018.21167
- Delamater PL, Street EJ, Leslie TF, et al. Complexity of the basic reproduction number (R0) - volume 25, number 1—January 2019 - emerging infectious diseases journal - CDC. Emerg Infect Dis. 2019;25:1–4. doi: 10.3201/eid2501.171901
- Bloom DE, Black S, Rappuoli R. Emerging infectious diseases: A proactive approach. Proc National Acad Sci. 2017;114:4055–4059. doi: 10.1073/pnas.1701410114
- Wolfe DN, Zarrabian AG, Disbrow GL, et al. Progress towards a vaccine against Ebola to meet emergency medical countermeasure needs. Vaccine. 2019;37:7178–7182. doi: 10.1016/j.vaccine.2017.10.111
- Broadbent AJ, Subbarao K. Influenza virus vaccines: lessons from the 2009 H1N1 pandemic. Curr Opin Virol. 2011;1:254–262. doi: 10.1016/j.coviro.2011.08.002
- Prevention C for DC and. Vaccine Testing and the Approval Process. (2020).
- Lurie N, Saville M, Hatchett R, et al. Developing covid-19 vaccines at pandemic speed. New Engl J Med. 2020;382:1969–1973. doi: 10.1056/NEJMp2005630
- Everts M, Cihlar T, Bostwick JR, et al. Accelerating drug development: antiviral therapies for emerging viruses as a model. Annu Rev Pharmacol. 2016;57:1–15.
- Cao B, Wang Y, Wen D, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. New Engl J Med. 2020;382:1787–1799. doi:10.1056/nejmoa2001282.
- Tu Y-F, Chien C-S, Yarmishyn AA, et al. A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci. 2020;21:2657. doi: 10.3390/ijms21072657
- Thorlund K, Dron L, Park J, et al. A real-time dashboard of clinical trials for COVID-19. Lancet Digital Heal. 2020;2:e286–e287. doi:10.1016/s2589-7500(20)30086-8.
- Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi:10.1016/s0140-6736(20)31022-9.
- Pelfrene E, Mura M, Sanches AC, et al. Monoclonal antibodies as anti-infective products: a promising future? Clin Microbiol Infec. 2018;25:60–64. doi: 10.1016/j.cmi.2018.04.024
- Salazar G, Zhang N, Fu T-M, et al. Antibody therapies for the prevention and treatment of viral infections. Npj Vaccines. 2017;2:19. doi: 10.1038/s41541-017-0019-3
- Marston HD, Paules CI, Fauci AS. Monoclonal antibodies for emerging infectious diseases — borrowing from history. New Engl J Med. 2018;378:1469–1472. doi: 10.1056/NEJMp1802256
- Froude JW, Stiles BG, Pelat T, et al. Antibodies for biodefense. Mabs. 2011;3:517–527. doi: 10.4161/mabs.3.6.17621
- McTamney P. Utilizing the human body as a bioreactor: direct comparison of in vivo expressed biologics (IVEB) delivery platforms. Antibodies as drugs: from B cell biology to new treatments; 2020 February; Santa Fe, NM; 2020.
- Wang X, Mathieu M, Brezski RJ. Igg Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9:63–73. doi: 10.1007/s13238-017-0473-8
- Smatti MK, Thani AAA, Yassine HM. Viral-induced enhanced disease illness. Front Microbiol. 2018;9:2991. doi: 10.3389/fmicb.2018.02991
- Tseng C-T, Sbrana E, Iwata-Yoshikawa N, et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. Plos One. 2012;7:e35421. doi: 10.1371/journal.pone.0035421
- Liu L, Wei Q, Lin Q, et al. Anti–spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. Jci Insight. 2019;4:e123158. doi: 10.1172/jci.insight.123158
- Hummel J, Pagkaliwangan M, Gjoka X, et al. Modeling the downstream processing of monoclonal antibodies reveals cost advantages for continuous methods for a broad range of manufacturing scales. Biotechnol J. 2019;14:1700665. doi: 10.1002/biot.201700665
- Shukla AA, Wolfe LS, Mostafa SS, et al. Evolving trends in mAb production processes. Bioeng Transl Medicine. 2017;2:58–69. doi: 10.1002/btm2.10061
- Kelley B. Developing therapeutic monoclonal antibodies at pandemic pace. Nat Biotechnol. 2020;38:540–545. doi: 10.1038/s41587-020-0512-5
- Hernandez I, Bott SW, Patel AS, et al. Pricing of monoclonal antibody therapies: higher if used for cancer? Am J Managed Care. 2018;24:109–112.
- Greenwood B. The contribution of vaccination to global health: past, present and future. Philos Trans R Soc B Biol Sci. 2014;369:20130433. doi: 10.1098/rstb.2013.0433
- Mahy M, Stover J, Stanecki K, et al. Estimating the impact of antiretroviral therapy: regional and global estimates of life-years gained among adults. Sex Transm Infect. 2010;86:ii67.
- Bedford J, Farrar J, Ihekweazu C, et al. A new twenty-first century science for effective epidemic response. Nature. 2019;575:130–136. doi: 10.1038/s41586-019-1717-y
- Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916
- Patel A, Bah MA, Weiner DB. In vivo delivery of nucleic acid-encoded monoclonal antibodies. Biodrugs. 2020;34:273–293. doi: 10.1007/s40259-020-00412-3
- Wang D, Tai PW, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discovery. 2019;18:358–378. doi:10.1038/s41573-019-0012-9.
- Priddy FH, Lewis DJM, Gelderblom HC, et al. Adeno-associated virus vectored immunoprophylaxis to prevent HIV in healthy adults: a phase 1 randomised controlled trial. Lancet Hiv. 2019;6:e230–e239. doi: 10.1016/S2352-3018(19)30003-7
- Casazza JP. Durable HIV-1 antibody production in humans after AAV8-mediated gene transfer. Conference on retroviruses and opportunistic infections; 2020 March; Boston, MA; 2020.
- Gardner MR, Fetzer I, Kattenhorn LM, et al. Anti-drug antibody responses impair prophylaxis mediated by AAV-delivered HIV-1 broadly neutralizing antibodies. Mol Ther. 2019;27:650–660. doi:10.1016/j.ymthe.2019.01.004.
- Martinez-Navio JM, Fuchs SP, Pantry SN, et al. Adeno-associated virus delivery of anti-HIV monoclonal antibodies can drive long-term virologic suppression. Immunity. 2019;50:567–575.e5. doi: 10.1016/j.immuni.2019.02.005
- Kowalski PS, Rudra A, Miao L, et al. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27:710–728. doi: 10.1016/j.ymthe.2019.02.012
- Schlake T, Thran M, Fiedler K, et al. Messenger RNA, a novel avenue to antibody therapy? Mol Ther. 2019;27:773–784. doi: 10.1016/j.ymthe.2019.03.002
- Thran M, Mukherjee J, Pönisch M, et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. Embo Mol Med. 2017;9:1434–1447. doi: 10.15252/emmm.201707678
- Pardi N, Secreto AJ, Shan X, et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat Commun. 2017;8:ncomms14630. doi: 10.1038/ncomms14630
- Sabnis S, Kumarasinghe ES, Salerno T, et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010
- Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol. 2011;23:421–429. doi: 10.1016/j.coi.2011.03.008
- Algazi AP, Bhatia S, Agarwala SS, et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann Oncol. 2020;31:532–540. doi: 10.1016/j.annonc.2019.12.008
- Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867
- Mir L, Bureau M, Gehl J, et al. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262
- Rizzuto G, Cappelletti M, Maione D, et al. Efficient and regulated erythropoietin production by naked DNA injection and muscle electroporation. Proc Natl Acad Sci. 1999;96:6417–6422. doi: 10.1073/pnas.96.11.6417
- Tjelle TE, Corthay A, Lunde E, et al. Monoclonal antibodies produced by muscle after plasmid injection and electroporation. Mol Ther. 2004;9:328–336. doi: 10.1016/j.ymthe.2003.12.007
- Perez N, Bigey P, Scherman D, et al. Regulatable systemic production of monoclonal antibodies by in vivo muscle electroporation. Genet Vaccines Ther. 2004;2:1–5. doi: 10.1186/1479-0556-2-2
- Rosazza C, Meglic S, Zumbusch A, et al. Gene electrotransfer: a mechanistic perspective. Curr Gene Ther. 2016;16:98–129. doi: 10.2174/1566523216666160331130040
- Khan AS, Smith LC, Abruzzese RV, et al. Optimization of electroporation parameters for the intramuscular delivery of plasmids in pigs. DNA Cell Biol. 2003;22:807–814. doi: 10.1089/104454903322625019
- Schertzer JD, Plant DR, Lynch GS. Optimizing plasmid-based gene transfer for investigating skeletal muscle structure and function. Mol Ther. 2006;13:795–803. doi: 10.1016/j.ymthe.2005.09.019
- Molnar MJ, Gilbert R, Lu Y, et al. Factors Influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol Ther. 2004;10:447–455. doi: 10.1016/j.ymthe.2004.06.642
- Bureau MF, Naimi S, Ibad TR, et al. Intramuscular plasmid DNA electrotransfer. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 2004;1676:138–148. doi: 10.1016/j.bbaexp.2003.11.005
- Demonbreun AR, McNally EM. DNA electroporation, isolation and Imaging of Myofibers. J Visualized Exp. 2015;106:e53551.
- Wang X-D, Tang J-G, Xie X, et al. A comprehensive study of optimal conditions for naked plasmid DNA transfer into skeletal muscle by electroporation. J Gene Med. 2005;7:1235–1245. doi: 10.1002/jgm.765
- Lesbordes J-CC, Bordet T, Haase G, et al. In vivo electrotransfer of the cardiotrophin-1 gene into skeletal muscle slows down progression of motor neuron degeneration in pmn mice. Hum Mol Genet. 2002;11:1615–1625. doi: 10.1093/hmg/11.14.1615
- Yamazaki T, Nagashima M, Ninomiya D, et al. Passive immune-prophylaxis against influenza virus infection by the expression of neutralizing anti-hemagglutinin monoclonal antibodies from plasmids. Jpn J Infect Dis. 2011;64:40–49.
- Flingai S, Plummer EM, Patel A, et al. Protection against dengue disease by synthetic nucleic acid antibody prophylaxis/immunotherapy. Sci Rep. 2015;5:12616. doi: 10.1038/srep12616
- Elliott ST, Kallewaard NL, Benjamin E, et al. DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. npj Vaccines. 2017;2:18. doi: 10.1038/s41541-017-0020-x
- Muthumani K, Block P, Flingai S, et al. Rapid and long-term immunity elicited by DNA-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J Infect Dis. 2016;214:369–378. doi: 10.1093/infdis/jiw111
- Andrews CD, Luo Y, Sun M, et al. In vivo production of monoclonal antibodies by gene transfer via electroporation protects against lethal influenza and Ebola infections. Mol Ther Methods Clin Dev. 2017;7:74–82. doi: 10.1016/j.omtm.2017.09.003
- Patel A, Park DH, Davis CW, et al. In vivo delivery of synthetic human DNA-encoded monoclonal antibodies protect against Ebolavirus infection in a mouse model. Cell Rep. 2018;25:1982–1993.e4. doi: 10.1016/j.celrep.2018.10.062
- Choi H, Kudchodkar SB, Reuschel EL, et al. Synthetic nucleic acid antibody prophylaxis confers rapid and durable protective immunity against Zika virus challenge. Hum Vaccin Immunother. 2019;16(4):907–918.
- Esquivel RN, Patel A, Kudchodkar SB, et al. In vivo delivery of a DNA-encoded monoclonal antibody protects non-human primates against Zika virus. Mol Ther. 2019;27:974–985. doi: 10.1016/j.ymthe.2019.03.005
- Liu Y, Cao W, Sun M, et al. AAABroadly neutralizing antibodies for HIV-1: efficacies, challenges and opportunities. Emerg Microbes Infec. 2020;9:194–206. doi: 10.1080/22221751.2020.1713707
- Caskey M. Broadly neutralizing antibodies for the treatment and prevention of HIV infection. Curr Opin Hiv Aids. 2020;15:49–55. doi: 10.1097/COH.0000000000000600
- Klein F, Halper-Stromberg A, Horwitz JA, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492:118–122. doi: 10.1038/nature11604
- Kong R, Louder MK, Wagh K, et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J Virol. 2015;89:2659–2671. doi: 10.1128/JVI.03136-14
- Wise MC, Xu Z, Tello-Ruiz E, et al. In vivo delivery of synthetic DNA-encoded antibodies induces broad HIV-1-neutralizing activity. J Clin Invest. 2019;130:827–837. doi: 10.1172/JCI132779
- Mulangu S, Dodd LE, Davey Jr RT, et al. A Randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381:2293–2303. doi: 10.1056/NEJMoa1910993
- Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11:2251. doi: 10.1038/s41467-020-16256-y
- Wrapp D, Vlieger DD, Corbett KS, et al. Structural basis for potent neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell. 2020;181:1004–1015.e15. doi:10.1016/j.cell.2020.04.031.
- Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infection. 2020;80:607–613. doi:10.1002/jmv.25897 doi: 10.1016/j.jinf.2020.03.037
- Giambenedetto SD, Ciccullo A, Borghetti A, et al. Off-label Use of tocilizumab in patients with SARS-CoV-2 infection. J Med Virol. 2020. doi:10.1002/jmv.25897.
- Cavalli G, Luca GD, Campochiaro C, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2:e325–e331. doi: 10.1016/S2665-9913(20)30127-2
- Luo P, Liu Y, Qiu L, et al. Tocilizumab treatment in COVID-19: A single center experience. J Med Virol. 2020;92:814–818. doi:10.1002/jmv.25801.
- Tjelle TE, Salte R, Mathiesen I, et al. A novel electroporation device for gene delivery in large animals and humans. Vaccine. 2006;24:4667–4670. doi: 10.1016/j.vaccine.2005.08.068
- Hollevoet K, Vleeschauwer SD, Smidt ED, et al. Bridging the clinical gap for DNA-based antibody therapy through translational studies in sheep. Hum Gene Ther. 2019;30:1431–1443. doi: 10.1089/hum.2019.128
- Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. Aaps J. 2012;14:296–302. doi: 10.1208/s12248-012-9340-y