
ABSTRACT
Global dissemination of ciprofloxacin-resistant Salmonella Kentucky has been observed over the past decades. In recent years, there have been reports of extended-spectrum β-lactamase (ESBL) producing S. Kentucky. Routine surveillance at the European Centre for Disease Prevention and Control (ECDC) detected cases with a ciprofloxacin-resistant S. Kentucky with the ESBL-gene blaCTX-M-14b. Ensuing research identified 78 cases in 2013–2018 in eight European countries. Compared to other S. Kentucky and non-typhoidal Salmonella infections, reported to the European Surveillance System, these cases were more likely to be elderly and to present urinary-tract infections. Bayesian time-scaled phylogeny on whole genome sequences of isolates from these cases and supplementary isolates from public sequence databases was used to infer the origin and spread of this clone. We dated the origin of the blaCTX-M-14b clone to approximately 2005 in Northern Africa, most likely in Egypt. The geographic origin predicted by the phylogenetic analysis is consistent with the patients’ travel history. Next to multiple introductions of the clone to Europe from Egypt, our analysis suggests that in some parts of Europe the clone might have formed a stable population, from which further spread has occurred. Comparative genomics indicated that the blaCTX-M-14b gene is present on the bacterial chromosome, within the type VI secretion system region. The blaCTX-M-14b gene is integrated downstream of the hcp1 gene, on a 2854 bp plasmid fragment containing also ISEcp1. This is the first report of a chromosomally integrated CTX-M gene in Salmonella spp. in Europe, previous studies having identified similar genes only on plasmids.
Introduction
Encompassing more than 2500 serovars and a wide genetic diversity, Salmonella spp. is the second most prevalent cause of food-borne human infections in Europe, with an average of approximately 95,000 laboratory-confirmed cases reported per year [Citation1] in the last decade. With various ecological and epidemiological patterns, the numerous serovars circulate in a broad range of animal reservoirs and environmental niches from where they can spread to humans [Citation2,Citation3]. The diverse environmental exposure patterns and genetic plasticity of the bacteria (including the possibility to host a variety of mobile genetic elements) has shaped an accumulation of antimicrobial resistance genes [Citation4]. This is an emerging public health problem as some of the Salmonella isolates have become multidrug resistant, displaying resistance to several of the antibiotics commonly used for treatment of salmonellosis. One of the serovars that have acquired a large number of antibiotic resistance genes, is S. enterica subsp. enterica serovar Kentucky (henceforth named S. Kentucky). Showing a steady number of ∼600 cases reported per year EU-wide [Citation1] and resistance to fluoroquinolones, extended-spectrum β-lactams, and in some cases even to carbapenems, S. Kentucky is considered a high priority resistant pathogen according to the WHO global priority list [Citation5].
In recent years, various studies shed light on the emergence, drivers, and potential threats of S. Kentucky in general and of sequence type (ST) 198 in particular. S. Kentucky ST 198 seems to be a versatile microorganism that adapts easily to selection pressure exerted by the use of antibiotics in various environments. It has thus evolved from absence of resistance before 1990 to increasing prevalence of ciprofloxacin-resistant isolates beginning of the 21 century (from 55% in 2007 to 88% in 2017 [Citation4,Citation6]) and steady accumulation of genetic elements conferring resistance to other antibiotic classes in the early years of this decade [Citation6–8]. A common explanation for the continuous selective pressure is the intensive use of antibiotics in livestock farming and human medicine. Constant globalization of trade and travel contribute further to the spread of resistant bacterial clones. In Europe, human infections with multidrug-resistant S. Kentucky ST198 used to be predominantly associated with travel to North Africa or South-East Asia, with especially Egypt being cited as the most probable source of these bacterial isolates [Citation6,Citation7,Citation9].
The accumulation of fluoroquinolones resistance in S. Kentucky ST198 has been driven by mutations in the gyrA and parC genes [Citation9], but also by lateral transfer of genetic elements, such as plasmids, and insertion of parts of these elements in the bacterial chromosome by recombination [Citation10,Citation11]. Previous studies indicate the circulation in Europe of S. Kentucky isolates with a configuration of three ciprofloxacin-resistance inducing mutations: Ser83Phe and Asp87Asn/Asp87Tyr in gyrA, and Ser80Ile in parC [Citation6,Citation7,Citation9]. Additional chromosomal resistance genes were found inserted in the bacterial genome at various positions in the Salmonella Genomic Island 1 (SGI1) by mediation of insertion sequence 26 (IS26). Occasionally, genes encoding carbapenemases, e.g. blaOXA-48, blaNDM-1, and extended-spectrum β-lactamases (ESBLs) were found e.g. blaCTX-M-1, particularly on IncC and IncI1 plasmids [Citation9].
Recent years have brought about a further increase in the incidence of cases with S. Kentucky ST198 acquired within Europe harbouring both multidrug-resistance (77% in 2018) and high-level ciprofloxacin resistance (89% in 2018) [Citation12,Citation13]. A few countries have reported cases of S. Kentucky with ESBL to ECDC in the routine surveillance data for 2016 [Citation13]. This triggered further investigations which led to the discovery of an emergent S. Kentucky clone in Europe – one that next to ciprofloxacin resistance also harbours blaCTX-M-14b, a type of ESBL gene that is new for this serovar. This study aimed at investigating the epidemiology, phylogenomics, and comparative genomics of this new ESBL-producing clone of S. Kentucky ST198. By analysing a set of clinical isolates and putting them in the broader context of internationally collected isolates, we try to trace back the origins of this emerging clone and highlight the threat it might pose in the future for public health.
Material and methods
Epidemiological data
Epidemiological information on cases with S. Kentucky blaCTX-M-14b was collected from EU/EEA countries via an “urgent inquiry” launched in the ECDC Epidemic Intelligence Information System for Food- and Waterborne Diseases and Zoonoses (EPIS-FWD) in March 2018. Nine EU/EEA countries had observed such cases in recent years, and of these, eight could participate in the study. Data were collected on age, gender, travel, specimen type and date of receipt of the isolate at the reference laboratory. The Fisher exact test was used to compare frequencies in gender, age groups and specimen types of cases with S. Kentucky blaCTX-M-14b with those of other S. Kentucky and of other non-typhoidal Salmonella reported with AMR data to the European Surveillance System (TESSy) at ECDC for the years 2013–2018. A significance level of 0.05 was applied.
Sequence dataset
Sequences from 78 clinical isolates and one food isolate of S. Kentucky blaCTX-M-14b as well as from 35 other S. Kentucky ST198 circulating in eight participant countries, were collected at ECDC. These 114 sequences were supplemented with 166 other S. Kentucky ST198 from various public sequence databases to provide context. These were selected based on a maximum 50 cgMLST loci dissimilarity with the blaCTX-M-14b isolates, using the Enterobase cgMLST schema and filtering options [Citation14]. The resultant dataset contained a total of 280 sequences, spanning 17 years (Table S1).
Bacterial isolates processing
The sequencing of the Salmonella isolates was performed on various Illumina platforms (Illumina, San Diego, CA, U.S.A.), i.e. HiSeq 2000, HiSeq 2500, and MiSeq PE300 with the appropriate Illumina library protocols. Paired-end FASTQ files were quality trimmed and de novo genome assemblies were made using CLC Genomics Workbench v 10.0 (Qiagen, Hilden, Germany). The parameters for trimming were as follows; ambiguous limit: 3, quality limit: 0.05. The parameters for the de novo assembly were as follows. For mapping settings; mapping mode: map reads back to contigs (slow), update contigs selected, mismatch cost: 2, insertion cost: 3, deletion cost: 3, length fraction: 0.5, similarity fraction: 0.8, match mode: random. For de novo settings; bubble size: 50, word size: 20, minimum contig length: 200 bp, perform scaffolding selected; auto-detect paired distances selected.
For identification of single nucleotide polymorphisms (SNPs) the short paired-end reads were mapped to a reference genome of S. Kentucky (GenBank accession number CP026327) using BWA-MEM [Citation15]. SAM files from this aligner were sorted and filtered into BAM files using SAMtools [Citation16]. The VCF files with high quality SNPs (>90% consensus, minimum depth 15x, GQ > = 20) were created using BCFtools [Citation16]. The SNPs present in regions potentially resulting from recombination processes were further identified using Gubbins v1 [Citation17] with default settings and subsequently removed.
For several isolates (S16BD08730, S18BD00684, S18BD03994, and S18BD05011), additional long-sequence reads were obtained at Sciensano with a MinION sequencer (Oxford Nanopore Technology), in view of a hybrid assembly. The respective isolates were considered to be good representatives of the diversity of S. Kentucky observed in our dataset. The long-read sequencing library was prepared with the 1D native barcoding genomic DNA (with EXP-NBD103 and SQK-LSK108 kits) protocol from Oxford Nanopore Technologies. The long-read sequencing library pool consisting of 12 barcodes (including other samples not relevant for this study) was loaded onto a 9.4 MinION flowcell (FLO-MIN106) and sequenced for 48 h. Local basecalling was performed with Guppy (v3.2.4) with the trimming option enabled that removes low quality bases before the adapter sequence. For the hybrid assembly the same software and settings as described previously [Citation18] were used. The hybrid assemblies were used to verify the genomic context of the blaCTX-M alleles.
Annotation
The overall gene content and position of genes on the genomic contigs of the isolates was assessed using the annotation tool Prokka v1 [Citation19], with CP026327 as reference genome. Alignment of genomic regions was performed using the BLAST feature of Easyfig [Citation20].
Genome mining
Genome assemblies were screened with BLAST+ v.2.9.0 [Citation21,Citation22] for the presence of plasmid replicons, antibiotic resistance genes, pathogenicity islands, and insertion sequences, using the PlasmidFinder [Citation23], ResFinder [Citation24], PAI [Citation25,Citation26], and ISfinder [Citation27,Citation28] databases (accessed at 30 November 2018), respectively. The thresholds for considering one of the genetic features present were 60% coverage and 90% identity with the target.
Phylogenetics and phylodynamics
For the phylogenetic analysis, we used only the SNPs present in the homologous regions of the genome. BEAST v.2.5.0 [Citation29] was used for the time calibrated Bayesian inference of phylogeny, where the calibrator was the sampling year of the isolates. The substitution model best fitting the data was selected to be TVM + Gamma, as assessed in jModelTest2 [Citation30,Citation31]. Due to a low temporal signal in the data (R2 of the heuristic residual mean squared = 0.1541), as inferred by the root-to-tip regression in TempEst v.1.5.1 [Citation32], a log-normal uncorrelated relaxed molecular clock was used. The choice for a specific demographic model was based on the number of population changes from the Extended Bayesian Skyline Plot analysis, with 108 steps and sampling every 5000 steps. Since the 95%HPD did not contain 0, indicating at least one change in the bacterial population size, we used a Bayesian Skyline demographic model, which was run three times, each with 1.5 × 108 steps and sampling every 10,000 steps. The maximum clade credibility tree was based on 34,000 trees.
Estimates of the demographic changes in the bacterial population were calculated for both the entire dataset and the blaCTX-M-14b clone using Tajima's D [Citation28] as implemented in the “pegas” package v.0.11 [Citation33] in R v.3.6.0 [Citation34].
The most likely country of origin for the various clades in the final BEAST phylogenetic tree was inferred using maximum likelihood estimation of discrete ancestral states with an equal rates model for state transition in the “ape” package v.5.3 [Citation35] in R.
Results
We have analysed European clinical isolates belonging to S. Kentucky ST198 blaCTX-M-14b for distribution, phylodynamics, and presence of molecular markers associated with antibiotic resistance. Next to the 78 CTX-M-14b human isolates, we have used one additional CTX-M-14b food isolate from peppermint imported from Egypt (provided by Public Health England), and 201 other S. Kentucky ST198 isolates collected from either countries participating in this inquiry (n = 35), or the public domain (n = 166) in order to put the findings into the broader context of internationally circulating bacteria, and to reliably reconstruct the phylogeny. Details on all isolates included in the study can be found in the supplementary material (Table S1). For clarity, through the rest of the manuscript, we will refer to the 78 CTX-M-14b isolates as the clinical dataset, and to the total number of isolates used in the study (clinical, food and public databases) as the extended dataset.
Epidemiology
Of the 78 S. Kentucky blaCTX-M-14b clinical cases included in the study, 24 were from the United Kingdom, 21 from Malta, 21 from the Netherlands, five from Germany, two each from Belgium, Denmark, and Norway and one from Ireland. The first case was identified in February 2013 and the last case in August 2018 (). Twenty cases had reported travel within the incubation period of which 11 to Egypt, eight to Malta and one with unknown destination. Forty-five cases (58%) were male and 33 (42%) female. The proportion of males among cases with S. Kentucky blaCTX-M-14b was not significantly different than those of other non-typhoidal Salmonella serovars, though higher than in other S. Kentucky infections (). The age ranged from 0 to 95 years with most cases (42%) in persons 65 years and older (). The proportion elderly (65+ years) was significantly higher among cases with S. Kentucky blaCTX-M-14b in comparison to other S. Kentucky and other non-typhoidal Salmonella serovars (). Isolation of the bacteria from urine (14.1%) was also significantly more common among these cases compared to other S. Kentucky and other non-typhoidal Salmonella serovars ().
Figure 1. Distribution of S. Kentucky blaCTX-M-14b cases by year and quarter. Each square in the figure represents one case; the outer colour indicates the country where the case was reported, while the inner colour indicates the most probable country of infection when the case had travelled during the incubation period.
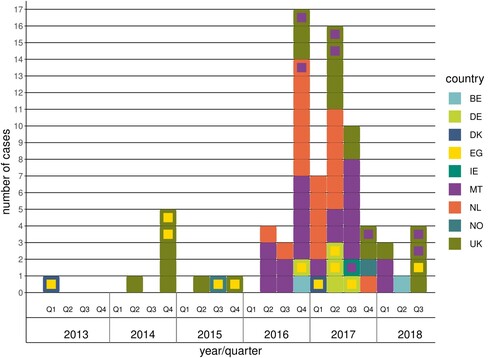
Table 1. Comparison of epidemiological data of the S. Kentucky CTX-M-14b with other S. Kentucky and non-typhoidal Salmonella.
Antibiotic resistance genes content
Genome mining for genes encoding for antimicrobial resistance indicated the presence of a wide spectrum of predicted resistance. Ten classes of antibiotics were included in the search list: aminoglycosides, β-lactams, fosfomycin, macrolides/lincosamides/streptogramin (MLS), phenicol, quinolones, rifampicin, sulphonamides, tetracycline, and trimethoprim. Resistance genes from all ten classes were found in the extended dataset (280 isolates), while in the clinical CTX-M-14b isolates (78 isolates) resistance genes from seven classes were found: aminoglycosides, β-lactams, fosfomycin, phenicol, sulphonamides, tetracycline, and trimethoprim. The majority of these latter isolates (74/78, 94.8%) presented resistance genes from four classes of antibiotics – aminoglycosides, β-lactams, sulphonamides, and tetracycline. Resistance to fosfomycin, phenicol, and trimethoprim was sporadic (Table S1). All 280 isolates harboured one or more aminoglycoside resistance gene, with 56.8% (159/280) of all isolates and 97.4% (76/78) of the CTX-M-14b isolates harbouring four or more aminoglycoside resistance genes, and 30.4% (85/280) of all isolates and 89.7% (70/78) of the CTX-M-14b isolates harbouring six or seven of these determinants (Table S1). The majority of them were associated with SGI1, as described previously [Citation9] i.e. aac(3)-Id, aadA7, aph(3″)-Ib, aph(3’)-Ia, and aph(6)-Id. The chromosomally located aac(6’)-Iaa, was present in all isolates.
The sul1 and tet(A) genes were also present in most of the CTX-M-14b isolates (94.8% or 78/78 and 96.1% or 75/78, respectively). Among all isolates investigated, these two genes were slightly less prevalent (80.0% or 224/280 and 86.8% or 243/280, respectively). Three isolates carried also other β-lactamase encoding genes: non-ESBL blaTEM variant (1.3% or 1/78), and carbapenemase blaOXA-48 (2.56% or 2/78; Table S1). In the extended dataset, the blaTEM gene was present in 51.8% (145/280) of the isolates, with a majority of blaTEM-1B (144/280), and one instance of blaTEM-1C (1/280), while blaOXA-48 was only demonstrated in the CTX-M-14b isolates (Table S1). The peppermint isolate presented a similar antibiotic resistance pattern to the clinical S. Kentucky blaCTX-M-14b isolates (Table S1).
Genes qnrB4 and qnrB19, conferring resistance to quinolones were present in only four isolates from the extended dataset (1.43% or 4/280), but not among the CTX-M-14b isolates. Point mutations involved in the development of ciprofloxacin resistance were identified in the gyrA and parC genes. The CTX-M-14b clinical isolates presented the nonsynonymous mutations Ser80Ile in ParC, and Asp87Gly in GyrA. Other isolates in the extended dataset presented also Asp87Asn (84 isolates), and Asp87Tyr (53 isolates) in GyrA (Align S1, Align S2). All clinical CTX-M-14b isolates were resistant to ciprofloxacin. From the extended dataset, only five were susceptible to ciprofloxacin (5/280 or 1.78%); these were isolated between 2001 and 2005, and presented the wildtype aminoacids in positions 87 of GyrA (D) and 80 of ParC (S).
The in silico characterization of the antibiotic resistance profile identified the S. Kentucky CTX-M-14b as a set of MDR isolates with molecular markers encoding for resistance to aminoglycosides, fluoroquinolones, β-lactams, sulphonamides, and tetracycline.
Plasmid replicon content
We have used the detection of plasmid replicons (short fragments of plasmid DNA responsible for the initiation and control of plasmid replication) to infer presence of certain plasmids in the bacterial isolates (as described in [Citation36]) i.e. we assume that if the replicon specific for a certain plasmid class is detected, the plasmid will be present in its entirety. The plasmid replicon content analysis revealed a wide range of plasmids in the extended dataset, with several small plasmids and multiple different Inc families encountered, of which Col(pVC) and IncI1 were the most abundant (22/280 or 7.86% and 10/280 or 3.57% isolates, respectively). The distribution of the plasmids in the clinical CTX-M-14b isolates reflected also the distribution of the plasmids in the extended dataset (Table S1). Many of the isolates (31/280 or 11.07%) contained one plasmid only. Two and more plasmid types were found in seven isolates (7/280 or 2.5%; Table S1).
Phylogenetics and phylodynamics
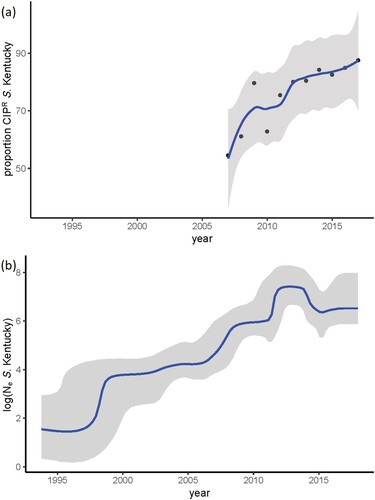
After extraction of the core genome SNPs and filtering of the high frequency variants, potentially indicative of recombination, a total number of 2650 SNPs remained to be used in the phylogenetic inference. Two Dutch CTX-M-14b clinical isolates from this study were identical in their SNP profile and consequently, one of them was discarded, resulting in a phylogeny of 279 terminal nodes. Phylogenetic reconstruction of the extended dataset indicated the presence of three major clades, corresponding roughly to the amino acid substitutions in GyrA (). The ciprofloxacin susceptible isolates clustered close to the root of the tree, which was estimated at ∼ 1993 (). The origin of the ciprofloxacin resistance (CIPR) was dated to 1998 (95%HPD 1995-1999). The reconstruction of ancestral states of the nodes in the phylogeny indicates Egypt as the most probable origin of CIPR, followed by spread all over the world (). Bayesian skyline analysis showed an increase in the effective population size of CIPR S. Kentucky ST198 ((b)), which is consistent with the statistics from TESSy on the increased prevalence of CIPR in Europe ((a)). Additionally, the negative Tajima's D (−2.72, p = 0.006) indicated a potential demographic expansion of CIPR S. Kentucky ST198.
Figure 2. Time-scaled phylogeny of S. Kentucky ST198. The coloured concentric rings represent, from the center outwards, the continent where the isolate was collected, the amino acid at position 87 in GyrA, and the amino acid at position 80 in ParC. Ciprofloxacin sensitive isolates are characterized by presence of D at position 87 of GyrA and of S at position 80 of ParC. The presence of black triangles immediately next to the tips of the tree indicate isolates that were collected from European countries participating in this inquiry. The squares in multiple concentric rings indicate the presence of a specific ESBL gene/allele (from the center outwards): blaCMY-2, blaCTX-M-104, blaCTX-M14b, blaCTX-M-15, and blaCTX-M-55.
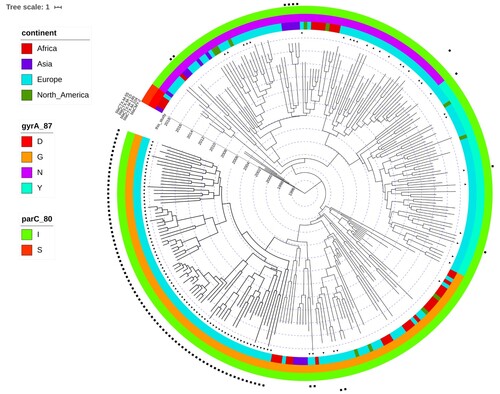
Figure 3. Temporal trends in S. Kentucky. (a) y-axis indicates the proportion of human cases with ciprofloxacin-resistant S. Kentucky among all S. Kentucky infections – data from ECDC; (b) Bayesian skyline plot of the extended dataset of S. Kentucky ST 198; y axis indicates the effective population size on a log10 scale.
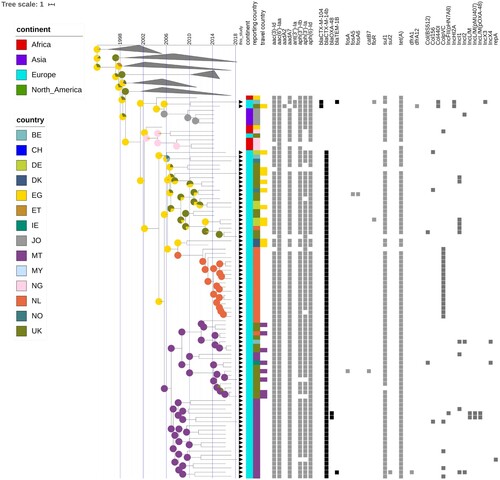
While resistance genes for cephalosporins are present in various branches of the phylogeny, the clone harbouring blaCTX-M-14b was contained within the Asp87Gly (G) clade. The CTX-M-14b isolate from peppermint imported from Egypt clustered with the rest of the CTX-M14b isolates. The genetic distance between blaCTX-M-14b and blaCTX-M-104, observed in a couple other isolates in an adjacent clade, is of only one point mutation – the adenine at position 824 is substituted by guanine (A824G), resulting in an amino acid change at codon 275 (Asn275Ser, Align S3, Align S4). Even with all additional isolates included in the analysis no human isolates carrying the blaCTX-M-14b gene could be found elsewhere outside of Europe (). However, isolates from ten blaCTX-M-14b positive persons have been associated to travel to Egypt. Some of the first introductions to Europe were to Germany and Norway, followed by almost concomitant introductions to Malta, UK and other EU countries. No less than eight isolates seem to have been acquired during travel to a European country – Malta (). The Bayesian reconstruction of phylogeny dated the most recent common ancestor (MRCA) of this clone to 2005 (95%HPD 2002-2006), and the reconstruction of the ancestral state indicates Egypt as its most likely country of origin (). The blaCTX-M-14b clade is structured in four clusters, rather homogeneous in terms of country of isolation. Remarkably, the isolates from the UK form two distinct clusters, heterogeneous in genetic diversity; one cluster is strongly associated with travel to Egypt, while the other one with travel to Malta (). The Dutch isolates form a low diversity cluster, and they coalesce with two isolates from Germany with travel record to Egypt. The cluster of isolates from Malta shows a large genetic diversity, with long branches sharing a MRCA in about 2007. The Bayesian skyline plot did not indicate an increase in effective population size of the blaCTX-M-14b clone. However, Tajima's D was < −2 (−2.38, p = 0.01), indicating a potential expansion of this clone.
Figure 4. Time-scaled phylogeny with epidemiological and molecular features of the S. Kentucky blaCTX-M-14b clade and closely related isolates. The presence of black triangles immediately next to the tips of the tree indicate isolates that were collected from European countries participating in this inquiry. The coloured strips indicate the continent, country of isolation, and country of infection for the respective isolates. The coloured squares indicate the presence of a specific antibiotic resistance gene or a plasmid. The pie charts associated with the internal nodes of the tree indicate the probability for the country of origin, with larger sectors of the circles corresponding to a higher probability for the country with the respective colour.
Genomic location of blaCTX-M14b
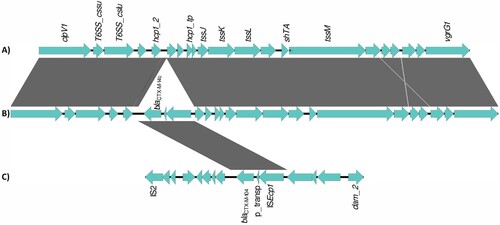
The architecture of the antibiotic resistance complement was complex, with multiple locations in the bacterial genome harbouring antibiotic resistance genes. Most genes encoding for resistance to cephalosporins (blaCTX-M and blaCMY) were located on plasmids, with the exception of blaCTX-M-14b which was always located on the chromosome. Genome alignment of isolates carrying this gene, with isolates from neighbouring branches of the phylogeny, and with reference isolates, showed that blaCTX-M-14b is present on a 2854 bp fragment inserted in the T6SS region of the bacterial genome, downstream of the hcp1 gene (position 3701330 of reference genome CP126327). The fragment is flanked on the one side by an insertion sequence – ISEcp1 from the IS1380 family of insertion sequences (). The 2854 bp fragment was absent on the chromosome of the non-blaCTX-M-14b. A similar construct was, however, found on plasmids in isolates paraphyletic to the CTX-M-14b clade; the only difference was that the respective plasmids carried another blaCTX-M allele - blaCTX-M-104, with one point mutation in comparison to blaCTX-M-14b (, Align S3, Align S4). Interestingly, one of the isolates carrying this ESBL gene on a plasmid had a travel record to Egypt.
Figure 5. Alignment of genomic segments from the chromosome and plasmid of representative S. Kentucky ST198 isolates. (A) chromosome of S. Kentucky isolate lacking the chromosomal blaCTX-M-14b, (B) chromosome of S. Kentucky isolate carrying the chromosomal blaCTX-M-14b, (C) plasmid of S. Kentucky isolate carrying blaCTX-M-104. Genomic loci are depicted as blue arrows and homology regions are depicted as grey blocks. The comparison of the T6SS regions on the bacterial chromosome of a blaCTX-M-14b negative isolate and a blaCTX-M-14b positive isolate show the insertion of a fragment carrying the blaCTX-M-14b gene, a putative transposase and the ISECp1 insertion sequence. This fragment is identical to a fragment located on the IncHIA2 plasmid of some blaCTX-M-14b negative isolates.
Discussion
At EU level, over the last years, the proportions of human Salmonella isolates resistant to the clinically important antimicrobials ciprofloxacin and cefotaxime were overall relatively low (12.5% and 1.5%, respectively in 2018)[Citation12]. However, in S. Kentucky, the seventh most common serovar in human Salmonella infections in 2018, an extremely high proportion (88.6%) of the isolates were high-level ciprofloxacin resistant [Citation12]. In addition, S. Kentucky shows relatively high levels of combined resistance to ciprofloxacin and cefotaxime. The highest proportion of S. Kentucky isolates with combined resistance was reported in Malta and related to ESBL-producing bacterial isolates [Citation4]. This triggered the current study aiming at reconstructing the time and place of emergence of this strain, carrying blaCTX-M-14b, which seems to have evolved from the high-level ciprofloxacin resistant, multidrug-resistant S. Kentucky ST198. The latter has spread rapidly throughout Europe and elsewhere in the world, both in humans and in the food chain, and is considered to originally have been imported to Europe via travellers to North Africa [Citation6]. To the date of completion of this analysis no ESBL-producing S. Kentucky carrying blaCTX-M-14b have been reported in food or animals to the European Food Safety Agency or the EU Reference Laboratory for antimicrobial resistance [Citation4].
The majority of European isolates of S. Kentucky ST198 used in this study contain multiple resistance genes to aminoglycosides. All of them contained mutations in the gyrA and parC genes, potentially involved in resistance to ciprofloxacin. Furthermore, ESBL-producing isolates in our dataset were not occasional, but amassed to more than 100 isolates, all collected in the past decade. Moreover, two of the CTX-M-14b isolates also contained a carbapenemase gene, i.e. blaOXA-48.
So far, blaCTX-M genes in European S. Kentucky have always been described on plasmids. There have been several reports of sequential integration of antimicrobial resistance genes at specific locations in the chromosome, such as the SGI1 [Citation7,Citation9]. Here, we report the integration of blaCTX-M-14b in the bacterial chromosome of S. Kentucky isolated from European patients. The similarity in structure of the inserted fragment with a corresponding fragment on the IncHI2 plasmid in isolates retrieved from other European countries, highlights the possibility that the integration has occurred as result of lateral gene transfer from this plasmid to the chromosome. One of the isolates carrying this fragment on the plasmid was isolated from a patient in the UK, but with a travel history to Egypt. Although the blaCTX-M allele on the plasmid was a different one – blaCTX-M-104 – the difference between the two CTX-M alleles is one base substitution. This might suggest a two-step event that has led to the integration of blaCTX-M-14b in the chromosome, with either order of the events possible: (a) point mutation leading to an amino acid substitution (Asn275Ser), and (b) integration in the chromosome, mediated by an IS element belonging to the IS1380 family; IS elements are known to drive the expression of blaCTX-M genes [Citation37]. ISEcp1-mediated integration events have also been described for blaCMY genes in Acinetobacter spp. [Citation38]. Alternatively, another source for the blaCTX-M-14b might be lateral gene transfer with Escherichia coli. CTX-M-14b is highly prevalent in E. coli [Citation39-41], and also present on IncHI2 plasmids [Citation40], in the same genetic background (i.e. flanked by the same insertion sequence) as in our S. Kentucky isolates. Furthermore, the two bacterial species often share environmental and biological niches where the horizontal gene transfer could occur [Citation42-44]. Recently, S. Kentucky with a chromosomally located blaCTX-M-14b gene has been reported in China [Citation45] in carcass washing water from a poultry slaughterhouse, and other events of integration of a blaCTX-M gene in the bacterial chromosome have been reported in Chinese isolates of S. Chester, S. Indiana, and S. Typhimurium [Citation46], converging to the scenario of Salmonella spp. evolving towards multidrug-resistant bacteria due to the increased use of antibiotics in intensive farming settings.
Travel history of the patients from which the blaCTX-M-14b isolates have been retrieved indicate that this clone is prevalent in Egypt and that occasional infection of travellers occurs. The Dutch isolates forming a low diversity cluster were all isolated from an outbreak of salmonellosis in a retirement home, and they coalesced with isolates from people with travel history to Egypt. Additionally, the only non-human isolate containing blaCTX-M-14b was retrieved from fresh peppermint imported from Egypt in 2015. Furthermore, the ancestral state reconstruction for the country of infection indicates Egypt as the most probable origin, not only for the blaCTX-M-14b clone, but also for the entire CIPR clone. What has once been a local problem has become of international concern, with the international travel of people and/or trade of food potentially playing a role in the dispersion of the MDR bacteria. Egypt and North Africa were also mentioned in previous research [Citation7,Citation47] as the origin of CIPR S. Kentucky ST198 and commonplace for acquiring antibiotic resistance genes, and it is believed that intensive usage of antibiotics in poultry industry and animal husbandry is the cause for the development of multidrug-resistance in Salmonella spp. [Citation6,Citation46]. Furthermore, the high prevalence of the blaCTX-M-14b clone in Malta, both indigenously and in people travelling to this country, coupled with the high genetic diversity within the Maltese isolates, indicates potential establishment of this clone in a European country. Contrary to previous studies, where the acquisition of various AMR plasmids remained an isolated event [Citation9], here we report the clonal expansion of a bacterium carrying resistance determinants for aminoglycosides, ESBLs, quinolones, sulphonamides, and tetracycline. Classical population genetics metrics showed a significant bacterial population expansion of this clone. The failure to detect the increase of bacterial population size using a time-scaled Bayesian approach might be related to the lack of temporal signal, as the isolates were collected over a relatively short time (2013-2018).
We hypothesize that one of the factors leading to the success of the blaCTX-M-14b clone is the integration of the ESBL resistance gene in the bacterial chromosome. Although plasmid-mediated antibiotic resistance is an undesired situation it is of less consequence than the chromosomal-mediated one. Plasmids can be acquired, but also lost in bacterial evolution; the reduction of antibiotic use that would reduce the selective pressure on bacteria might arguably lead to elimination of plasmids from the population [Citation48,Citation49]. The ecological implication of the chromosomal-mediated antibiotic resistance is much more severe, as it does not allow the reversibility of the phenomenon – once the genes are integrated in the chromosome they are most likely to be maintained indefinitely, especially if they incur no fitness cost for the bacteria.
Although the main transmission mode of Salmonella spp. is zoonotic, commonly via contaminated food, the scarcity of non-human S. Kentucky blaCTX-M-14b isolates makes it difficult to assess what is its main source of transmission. While the main host of S. Kentucky is poultry, to the date when this analysis was performed, there had been no ESBL producing S. Kentucky isolated from poultry in European countries [Citation4]. The one non-human blaCTX-M-14b S. Kentucky isolate, identified in the UK, came from peppermint imported from Egypt, and it suggests a spill over of bacteria in the environment – from where they might end up on various greens via, e.g. irrigation water. A recent publication of ECDC and EFSA revealed that in 2018 there were six reports of cefotaxime resistant S. Kentucky isolates in poultry in three European countries: one from laying hens in Hungary, and five from broilers in the Netherlands and Malta (one and four respectively)[Citation12]. While all five broiler isolates were presumptive ESBL-producers, the genotypes and the genomic sequences are not known; we can therefore not make any allegations on the circulation or persistence of CTX-M-14b in poultry in Europe.
In 2016 and 2017, the cases from the Netherlands and Malta accounted for 2/3 of all S. Kentucky CTX-M-14b human cases. The vast majority of Dutch cases were associated with an outbreak in a retirement home while the Maltese cases were sporadic. In Malta, most of the elderly cases and one young adult had severe underlying comorbidities. Two Maltese cases developed symptoms while in hospital. Most cases were treated with antimicrobials until symptoms stopped, though a few became carriers over several months with repeated antimicrobial treatment. The significance of the high proportion of elderly and of urinary-tract infections among cases with S. Kentucky blaCTX-M-14b compared to other infections with S. Kentucky or other non-typhoidal Salmonella is unclear. It could indicate that the bacterium is more widespread in the community, with symptomatic cases being identified mainly among elderly and/or persons with immunosuppression/underlying disease. It could, however, indicate nosocomial transmission, i.e. transmission occurring in a hospital or health-care setting, where the antimicrobial pressure is high and where resistance genes could be shared between different bacterial species within the Enterobacteriaceae via horizontal gene transfer within and/or between patients [Citation50,Citation51]. For Salmonella, such transfer has been suggested for the finding of OXA-48 in MDR S. Kentucky in North Africa [Citation52]. Recently, a source attribution study regarding community-acquired ESBL-producing E. coli revealed humans as the most important source [Citation53]. Similarly, diarrhoea-causing infections with Shiga toxin-producing E. coli carrying the stx2f variant are hypothesized to have originated from a human reservoir [Citation54].
In this line, of increased recognition of humans as reservoir for pathogens previously thought to be strictly zoonotic, it could be hypothesized that S. Kentucky CTX-M-14b spreads from humans to humans. Hygiene and responsible use of antibiotics in health care and veterinary medicine remain important pillars of prevention. Since new cases of human infections with S. Kentucky CTX-M-14b continued being reported to national health authorities across Europe after the compilation of the dataset for this analysis, it seems that this clone is persistent in at least some parts of Europe. Investigating whether the persistence is driven by long term carriage of the bacteria in humans, or maintenance in animal reservoirs, requires continuous monitoring in both human and non-human populations in a One Health fashion. Further monitoring of temporal and spatial trends in the incidence of this clone, might assist in identification of potential non-human reservoirs, and possibly prevent establishment of multidrug-resistant bacteria in new geographical regions.
Authors’ contributions
P.J. Ceyssens, M.L. Borg, M. Chattaway, J. McCormick, T.J. Dallman, and E. Franz contributed with isolate sequences and epidemiological information on the human cases. A.H.A.M. van Hoek performed genome assemblies and SNP calling, and provided expertise for the interpretation of the of antibiotic resistance and plasmid profiles. S.C.J. de Keersmaecker and B. Berbers performed the MinION sequencing and hybrid assemblies. T. Westrell analysed the epidemiological data. C.E. Coipan performed genomics, phylodynamics, and statistical analyses. C.E. Coipan and T. Westrell wrote the manuscript. All authors reviewed and approved the manuscript.
Suppelemental_files.zip
Download ()Acknowledgements
The authors would like to thank the following persons for providing epidemiological and sequencing data on S. Kentucky isolates to this study: Mia Torpdahl and Kristoffer Kiil, Denmark; Sandra Simon and Gerhard Falkenhorst, Germany; Niall Delappe and Patricia Garvey, Ireland; Joël Mossong, Luxembourg; Umaer Naseer, Norway and Cecilia Jernberg, Sweden. We would also like to thank Rene Hendriksen, EURL-antimicrobial resistance, Kirsten Mooijman, EURL-Salmonella and Beatriz Guerra, EFSA for investigating their databases for matching food and animal isolates. Lastly, Chantal Quinten, ECDC, is acknowledged for statistical support on the epidemiological analysis and Johanna Takkinen, ECDC, for reviewing the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The accession numbers of the WGS for the isolates included in this study can be found in Table S1.
Related Research Data
References
- EFSA, ECDC. The European Union One health 2018 Zoonoses report. EFSA J.. 2019;17(12):e05926.
- Mughini-Gras L, van Pelt W. Salmonella source attribution based on microbial subtyping: does including data on food consumption matter? Int J Food Microbiol. 2014 Nov 17;191:109–115.
- Franz E, van Bruggen AH. Ecology of E. coli O157:H7 and Salmonella enterica in the primary vegetable production chain. Crit Rev Microbiol. 2008;34(3-4):143–161.
- EFSA, ECDC. The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017. EFSA J. 2019;17(2):e05598.
- WHO. Critically important antimicrobials for human medicine, 6th revision: WHO; 2019. Available from: https://apps.who.int/iris/bitstream/handle/10665/312266/9789241515528-eng.pdf?ua=1.
- Le Hello S, Harrois D, Bouchrif B, et al. Highly drug-resistant Salmonella enterica serotype Kentucky ST198-X1: a microbiological study. Lancet Infect Dis. 2013 Aug;13(8):672–679.
- Le Hello S, Hendriksen RS, Doublet B, et al. International spread of an epidemic population of Salmonella enterica serotype Kentucky ST198 resistant to ciprofloxacin. J Infect Dis. 2011 Sep 1;204(5):675–684.
- Le Hello S, Weill FX, Guibert V, et al. Early strains of multidrug-resistant Salmonella enterica serovar Kentucky sequence type 198 from Southeast Asia harbor Salmonella genomic island 1-J variants with a novel insertion sequence. Antimicrob Agents Chemother. 2012 Oct;56(10):5096–5102.
- Hawkey J, Le Hello S, Doublet B, et al. Global phylogenomics of multidrug-resistant Salmonella enterica serotype Kentucky ST198. Microb Genom. 2019 Jul;5(7):e000269.
- Fricke WF, McDermott PF, Mammel MK, et al. Antimicrobial resistance-conferring plasmids with similarity to virulence plasmids from avian pathogenic Escherichia coli strains in Salmonella enterica serovar Kentucky isolates from poultry. Appl Environ Microbiol. 2009 Sep;75(18):5963–5971.
- Johnson TJ, Thorsness JL, Anderson CP, et al. Horizontal gene transfer of a ColV plasmid has resulted in a dominant avian clonal type of Salmonella enterica serovar Kentucky. PLoS One. 2010 Dec 22;5(12):e15524.
- EFSA, ECDC. The European Union Summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020;18(3):e06007.
- EFSA, ECDC. The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2016. EFSA J. 2018;16(2):e05182.
- Zhou Z, Alikhan NF, Mohamed K, et al. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020 Jan;30(1):138–152.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–wheeler transform. Bioinformatics. 2009;25(14):1754–1760.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079.
- Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014;43(3):e15–e15.
- Berbers B, Saltykova A, Garcia-Graells C, et al. Combining short and long read sequencing to characterize antimicrobial resistance genes on plasmids applied to an unauthorized genetically modified Bacillus. Sci Rep. 2020 Mar 9;10(1):4310.
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069.
- Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410.
- Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
- PlasmidFinder db [Internet]. https://bitbucket.org/genomicepidemiology/plasmidfinder_db: 2019. Available from: https://bitbucket.org/genomicepidemiology/plasmidfinder_db.
- ResFinder [Internet]. https://bitbucket.org/genomicepidemiology/resfinder_db: 2019. Available from: https://bitbucket.org/genomicepidemiology/resfinder_db.
- PAIDB v2.0 [Internet]. http://www.paidb.re.kr/about_paidb.php: 2020.
- Yoon SH, Park YK, Kim JF. PAIDB v2.0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015 Jan;43(Database issue):D624–D630.
- ISfinder [Internet]. https://github.com/thanhleviet/ISfinder-sequences: http://www-is.biotoul.fr. 2018. Available from: https://github.com/thanhleviet/ISfinder-sequences.
- Siguier P, Perochon J, Lestrade L, et al. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34(suppl_1):D32–D36.
- Bouckaert R, Vaughan TG, Barido-Sottani J, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15(4):e1006650.
- Darriba D, Taboada GL, Doallo R, et al. Jmodeltest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704.
- Rambaut A, Lam TT, Max Carvalho L, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2(1):vew007.
- Paradis E. Pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics. 2010;26(3):419–420.
- RCoreTeam. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2017.
- Paradis E. Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2018;35(3):526–528.
- Carattoli A, Zankari E, Garcia-Fernandez A, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014 Jul;58(7):3895–3903.
- Vandecraen J, Chandler M, Aertsen A, et al. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit Rev Microbiol. 2017;43(6):709–730.
- Martínez T, Vázquez GJ, Aquino EE, et al. ISEcp1-mediated transposition of blaKPC into the chromosome of a clinical isolate of Acinetobacter baumannii from Puerto Rico. J Med Microbiol. 2014;63(12):1644–1648.
- Miro E, Mirelis B, Navarro F, et al. Surveillance of extended-spectrum beta-lactamases from clinical samples and faecal carriers in Barcelona, Spain. J Antimicrob Chemother. 2005 Dec;56(6):1152–1155.
- Navarro F, Mesa R-J, Miró E, et al. Evidence for convergent evolution of CTX-M-14 ESBL in Escherichia coli and its prevalence. FEMS Microbiol Lett. 2007;273(1):120–123.
- Bevan ER, Jones AM, Hawkey PM. Global epidemiology of CTX-M beta-lactamases: temporal and geographical shifts in genotype. J Antimicrob Chemother. 2017 Aug 1;72(8):2145–2155.
- Winfield MD, Groisman EA. Role of nonhost environments in the lifestyles of Salmonella and Escherichia coli. Appl Environ Microbiol. 2003;69(7):3687–3694.
- Lam LH, Monack DM. Intraspecies competition for niches in the distal gut dictate transmission during persistent Salmonella infection. PLoS Pathog. 2014;10(12):e1004527.
- Karberg KA, Olsen GJ, Davis JJ. Similarity of genes horizontally acquired by Escherichia coli and Salmonella enterica is evidence of a supraspecies pangenome. PNAS. 2011;108(50):20154–20159.
- Lei C-W, Zhang Y, Wang X-C, et al. Draft genome sequence of a multidrug-resistant Salmonella enterica serotype Kentucky ST198 with chromosomal integration of blaCTX-M-14b isolated from a poultry slaughterhouse in China. J Glob Antimicrob Resist. 2020;20:145–146.
- Zhang C-Z, Ding X-M, Lin X-L, et al. The emergence of chromosomally located blaCTX-M-55 in Salmonella from Foodborne animals in China [Original research]. Front Microbiol. 2019;10(1268).
- Le Hello S, Bekhit A, Granier SA, et al. The global establishment of a highly-fluoroquinolone resistant Salmonella enterica serotype Kentucky ST198 strain. Front Microbiol. 2013;4(395).
- De Gelder L, Ponciano JM, Joyce P, et al. Stability of a promiscuous plasmid in different hosts: no guarantee for a long-term relationship. Microbiology. 2007;153(2):452–463.
- Wein T, Hülter NF, Mizrahi I, et al. Emergence of plasmid stability under non-selective conditions maintains antibiotic resistance. Nat Commun. 2019;10(1):2595.
- Bosch T, Lutgens SPM, Hermans MHA, et al. Outbreak of NDM-1-producing Klebsiella pneumoniae in a Dutch hospital, with Interspecies transfer of the resistance plasmid and Unexpected Occurrence in Unrelated health care Centers. J Clin Microbiol. 2017 Aug;55(8):2380–2390.
- Torres-Gonzalez P, Cervera-Hernandez ME, Niembro-Ortega MD, et al. Factors associated to prevalence and incidence of Carbapenem-resistant Enterobacteriaceae Fecal carriage: A Cohort study in a Mexican Tertiary care hospital. PLoS One. 2015;10(10):e0139883.
- Ktari S, Le Hello S, Ksibi B, et al. Carbapenemase-producing Salmonella enterica serotype Kentucky ST198, North Africa. J Antimicrob Chemother. 2015 Dec;70(12):3405–3407.
- Mughini-Gras L, Dorado-Garcia A, van Duijkeren E, et al. Attributable sources of community-acquired carriage of Escherichia coli containing beta-lactam antibiotic resistance genes: a population-based modelling study. Lancet Planet Health. 2019 Aug;3(8):e357–e369.
- van Hoek A, van Veldhuizen JNJ, Friesema I, et al. Comparative genomics reveals a lack of evidence for pigeons as a main source of stx2f-carrying Escherichia coli causing disease in humans and the common existence of hybrid Shiga toxin-producing and enteropathogenic E. coli pathotypes. BMC Genomics. 2019 Apr 5;20(1):271.