
ABSTRACT
The MERS-CoV isolated during the 2015 nosocomial outbreak in Korea showed distinctive differences in mortality and transmission patterns compared to the prototype MERS-CoV EMC strain belonging to clade A. We established a BAC-based reverse genetics system for a Korean isolate of MERS-CoV KNIH002 in the clade B phylogenetically far from the EMC strain, and generated a recombinant MERS-CoV expressing red fluorescent protein. The virus rescued from the infectious clone and KNIH002 strain displayed growth attenuation compared to the EMC strain. Consecutive passages of the rescued virus rapidly generated various ORF5 variants, highlighting its genetic instability and calling for caution in the use of repeatedly passaged virus in pathogenesis studies and for evaluation of control measures against MERS-CoV. The infectious clone for the KNIH002 in contemporary epidemic clade B would be useful for better understanding of a functional link between molecular evolution and pathophysiology of MERS-CoV by comparative studies with EMC strain.
Introduction
A decade after the outbreak of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002, another highly pathogenic human CoV Middle East respiratory syndrome (MERS) CoV emerged in two different cities in the Arabian Peninsula region, Zarqa, Jordan in April 2012 [Citation1] and Jeddah, Saudi Arabia in June 2012 [Citation2]. Since the first reported case of MERS and the epidemics are still ongoing [Citation3]. As of January 2020, there were a total of 2,519 laboratory-confirmed cases of MERS-CoV with 866 casualties in 27 countries (case-fatality rate: 34.4%) [Citation4,Citation5].
Besides the majority of cases reported in Saudi Arabia, the largest outbreak outside of the Arabian Peninsula occurred in May 2015 in South Korea [Citation6–8]. The outbreak in South Korea was distinctive in clinical, pathological, and epidemiological aspects compared with the Saudi Arabian epidemic. For a half year, MERS-CoV infected 186 individuals and caused 38 deaths [Citation9]. Such a substantially lower fatality (∼ 20%) as compared with very early cases of MERS-CoV infection in Saudi Arabia in 2012–2013 [Citation10] might be attributed to differences in virulence and/or transmissibility between Korean isolates and earlier isolates of MERS-CoV, including the EMC strain (EMC/2012) identified by the Erasmus Medical Center in Amsterdam, Netherlands [Citation2].
The outbreak in South Korea was developed mainly via nosocomial transmission in which a single patient from Saudi Arabia initiated human-to-human spread throughout 17 hospitals [Citation6,Citation7]. The wide-spreading via interhuman transmission was mainly caused by three super-spreading events (via the index patient, Patient #1, and two other super-spreaders, Patient #14 and Patient #16 [Citation6]; see also A). By contrast, during a similar human-to-human transmission within a health care facility in Saudi Arabia [Citation10], the virus did not show this atypical wide interhuman spreading profile, according to the basic reproduction number (R0) estimated by Bayesian analysis [Citation11]. It has been suspected that pathogen-derived factors might account for such a unique spreading profile of the virus during the 2015 outbreak in South Korea [Citation6,Citation7]. In fact, previous studies proposed, albeit controversial, potential roles of spike (S) protein variants in wide interhuman MERS-CoV spreadings in Korea [Citation12–14]. Nevertheless, it is yet unknown which viral genetic variations prompted consecutive human-to-human transmissions (at least 5 rounds of transmissions) during the 2015 outbreak.
Figure 1. Construction of a full-length cDNA clone of a MERS-CoV Korean isolate. (A) Transmission routes and the origin of the MERS-CoV isolate used for the construction of an infectious cDNA clone in this study. The index patient (Patient #1) acquired the virus from an unknown source during his visit to the Middle East in 2015. Patient #1 spread the virus via nosocomial routes and also transmitted the virus to his spouse (Patient #2) in a household setting. A sputum sample from Patient #2 was inoculated onto Vero E6 cells, and viruses were passaged three times prior to sequencing analysis of the viral genome (KT029139). The purified virus (KNIH002 strain) was further passaged three times in Huh7 cells at the Korea Research Institute of Chemical Technology (Daejeon, Korea). Total RNA extracted from culture supernatants was then used to construct a full-length cDNA clone of the MERS-CoV Korean isolate. (B) Schematic illustration of the full-length cDNA clone of KNIH002. The cassette vector pBAC-MERS-CoV/YKC-5′-3′ carries a CMV promoter fused to the cDNA representing the first 811-nt of the genome, a multiple cloning site (MCS), and the cDNA of the last 950-nt (nt 29,159–30,108) of the genome followed by poly(dA)28, HDV ribozyme (Rz), and bovine growth hormone transcription termination and polyadenylation signal (BGH). The four unique restriction enzyme sites within MCS were used for the assembly of cDNA fragments (M-CoV-1, M-CoV-2, M-CoV-3, and M-CoV-4). Three nonsynonymous mutations incorporated in the resulting full-length clone (pBAC-MERS-CoV/YKC) are shown in red below the genome.
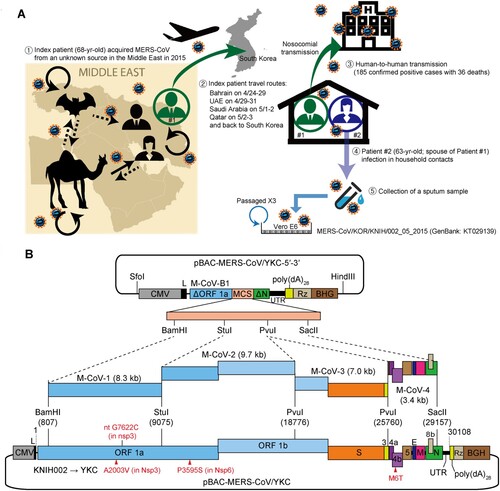
Reverse genetics systems for RNA viruses provide great tools for functional analysis of viral genes and development of antivirals and vaccines [Citation15]. The currently available systems established by different methodologies were all for the MERS-CoV EMC strain (JX869059) [Citation16–18], which is the prototype clade A strain isolated in 2012 [Citation2]. The clade A is currently composed of less than 10 isolates including another human isolate from Jordan (Jordan-N3/2012) and a few strains isolated from dromedaries [Citation19]. The MERS-CoV Korean isolate KNIH002 (KT029139) and all other strains isolated from humans during the 2015 outbreak in South Korea belong to the contemporary epidemic clade B that is is phylogenetically far from the clade A where the prototype MERS-CoV EMC strain belongs to [Citation2,Citation8,Citation19].
In the present study, we established a bacterial artificial chromosome (BAC)-based reverse genetics system for the MERS-CoV KNIH002 strain to better understand a functional link between molecular evolution and pathophysiology of MERS-CoV by comparative studies with the EMC strain. Using the recombinant virus recovered from the infectious cDNA clone, we compared the growth kinetics of the rescued virus with its parental strain and EMC strain. We also provide evidence of genetic instability of ORF5, frequently encountered during serial passages of MERS-CoV in vitro. The results from our analysis of the infectious clone recapitulate rapid molecular evolution of MERS-CoV in laboratory settings as well as its natural reservoirs, highlighting the importance of the use of a low-passage virus strain or a virus stock rescued from an infections clone to minimize the effect of nonidentified variants.
Materials and methods
Cell culture and virus infection
Baby hamster kidney cells (BHK-21), human hepatocellular carcinoma Huh7 cells, and African green monkey kidney-derived Vero E6 cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 µg/ml streptomycin at 37°C in 5% CO2. All cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The EMC strain was kindly provided by Bart Haagmans (Department of Viroscience, Erasmus Medical Center, Rotterdam, the Netherlands). The KNIH002 strain of MERS-CoV was received from the Korea Centers for Disease Control & Prevention (KCDC). The KNIH002 viral stock was propagated in Vero E6 cells grown in minimal essential medium (MEM) containing 2% FBS. Recombinant MERS-CoVs recovered from infectious cDNA clones were propagated in Huh7 cells grown in DMEM containing 10% FBS. For virus infection, cells were seeded in T25 flasks, cultured overnight, and then infected with viruses at an MOI of 0.001 by incubating at 37°C for 1 h. After washing, the infected cells were maintained in a complete medium supplemented with 2% FBS for the indicated periods as specified in figure legends. All experiments with MERS-CoVs were conducted within a biosafety level 3 (BSL3) facility at the KCDC.
MERS-CoV cDNA synthesis and sequencing analysis
Total RNA was extracted from the culture supernatant of MERS-CoV/KNIH002-infected cells using TRIzol LS reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using the SuperScript III First-Strand Synthesis System (Invitrogen) and PCR-amplified using specific primer sets (). The resulting PCR products were gel-purified using the QIAEX II gel extraction kit (Qiagen, Valencia, CA, USA) and cloned into pMW119 (Nippon Gene, Tokyo, Japan). Following sequencing of cDNA clones, overlapping sequences were assembled and compared with reference sequences using the AlignX program (Vector NTI Advance 10, Invitrogen). When indicated, specific regions of the viral genome (1.5–2.5 kb) were RT–PCR-amplified and cloned into pCR2.1-TOPO vector (Thermo Fisher Scientific, Waltham, MA, USA) to analyse cell culture-adapted mutations. When required, cDNAs containing a detrimental deletion or a point mutation were repaired using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA).
Table 1. Primers used to generate MERS-CoV cDNA fragments
Construction of a full-length cDNA clone of MERS-CoV
A full-length cDNA clone of MERS-CoV Korean isolate was constructed according to the protocol described previously [Citation16,Citation20]. The backbone cassette vector pBAC-MERS-CoV/YKC-5′-3′ was generated using the SARS-CoV subgenomic replicon pSARS-REP-Feo [Citation21] by assembling the cDNA M-CoV-B1 (representing the 5′-end region of MERS-CoV) and the chemically synthesized DNA fragment M-CoV-B2 (Bioneer, Daejeon, South Korea) using the In-Fusion HD Cloning Kit (Takara Bio, Tokyo, Japan). The M-CoV-B2 was composed of a multiple-cloning site (MCS containing BamHI, StuI, PvuI, and SacII sites) and the 950-bp long 3′-terminal sequences representing the nt 29,159–30,108 of viral genome plus a 28-bp long poly(A) tail and hepatitis delta virus (HDV) ribozyme. Based on the sequence of MERS-CoV KNIH002 (KT029139) [Citation8], four selected restriction enzymes, BamHI (nt 807), StuI (nt 9,072), PvuI (nt 18,776 and 25,760), and SacII (nt 29,157) were used for the assembly of cDNA fragments. A single nt change G7622C was made in nonstructural protein 3 (nsp3) gene to eliminate a StuI restriction site in the M-CoV-1 cDNA fragment. This substitution was also used as a genetic marker of the resulting full-length cDNA clone.
Then the four overlapping cDNA fragments flanked by the selected restriction sites (M-CoV-1 to M-CoV-4) were sequentially cloned into the MCS of pBAC-MERS-CoV/YKC-5′-3′ to generate pBAC-MERS-CoV/YKC, a full-length cDNA clone of the Korean isolate of MERS-CoV KNIH002. The genetic integrity of the final full-length cDNA clone was verified by sequencing. All the works using cDNAs of MERS-CoV were approved by KCDC (16-RDM-010) and by the Institutional Biosafety Committee at Yonsei University (Permit No: IBC-A-201605-117-02).
Construction of a recombinant MERS-CoV cDNA clone expressing RFP
The pMW119-M-CoV-4 containing the M-CoV-4 cDNA described above was PCR-amplified using the forward primer M-CoV-4 27512 F (5′-TAAGCAGCTCTGCGCTACTATGG-3′) and the reverse primer M-CoV-4 26842 R (5′-CATAGTTCGTTAAAATCCTGGATGATG-3′) to generate a linearized cassette vector. The RFP gene was PCR-amplified using ptdTomato-N1 plasmid (Takara Bio) as a template and the forward primer tdT 26825F (5′-GGATTTTAACGAACTATGGTGAGCAAGGGCGAGGAGG-3′) and the reverse primer tdT 27529R (5′-GTAGCGCAGAGCTGCTTACTTGTACAGCTCGTCCATGCC-3′). After gel purification, the RFP gene was cloned into the cassette vector using the In-Fusion HD Cloning Kit (Takara Bio). The resulting cDNA fragment carrying RFP gene, M-CoV-4-RFP, was then inserted into PvuI/SacII-digested pBAC-MERS-CoV/YKC to construct pBAC-MERS-CoV/YKC-RFP.
Rescue of recombinant viruses from infectious cDNA clones
Recombinant MERS-CoVs were recovered as previously described [Citation16,Citation20]. Briefly, 5 × 105 BHK-21 cells plated on a 6-well plate the day before transfection were cultivated in the absence of antibiotics overnight and were transfected with 5 µg of pBAC-MERS-CoV/YKC or pBAC-MERS-CoV/YKC-RFP pre-incubated with 12 µl of Lipofectamine 2000 transfection reagent (Invitrogen). Following incubation for 6 h, cells were then washed and detached by trypsin treatment, and resuspended cells were plated over confluent Huh7 monolayers grown in a T25 flask. The co-culture was incubated until clear signs of cytopathic effects (CPE) were observed, and the supernatant was harvested by clearing cell debris by centrifugation to obtain P0 viral stock. The P0 was passaged on fresh cells to generate P1, which was used in infection experiments unless otherwise specified.
Plaque assay
Viral titre was determined by plaque-forming assay as previously reported [Citation22]. Briefly, Vero E6 cells were seeded in a 6-well plate, cultured overnight, and infected with 10-fold serially diluted virus samples in a serum-free medium for 1 h with gentle rocking every 15 min. After washing with PBS, cells were overlaid with DMEM containing 1% low-melting agarose (Sigma-Aldrich, St. Louis, MO, USA), 2% FBS, 100 U/ml of penicillin, and 100 µg/ml streptomycin. After incubation for 3–4 days until clear plaques were observed, cells were fixed with 10% formaldehyde and stained with 1% crystal violet solution.
Real-time reverse-transcription quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. The MERS-CoV subgenomic RNA (sgRNA) copy number was determined as previously described [Citation16,Citation23] with minor modifications. Briefly, total RNA (500 ng) was subjected to cDNA synthesis using ImProm-II Reverse Transcriptase (Promega, Madison, WI, USA). Real-time RT-qPCR was carried out in triplicate using iQ Supermix (Bio-Rad, Hercules, CA, USA) and a set of primers [a forward primer M-CoV_sgRNA-N_28F (5′-ACTTCCCCTCGTTCTCTTGCAG-3′), a reverse primer M-CoV_sgRNA-N_28732_R (5′- GTAAGAGGGACTTTCCCGTGTTG-3′), and a TaqMan probe M-CoV_sgRNA-rtP (5′-FAM- CACGAGCTGCACCAAATAACACTGTCTC-3′-BHQ)] on a Bio-Rad CFX real-time PCR detection system (Bio-Rad). Standard RNA was prepared by in vitro transcription using a T7 promoter-fused DNA template representing nt 1–67 plus nt 28,545–28,746 [leader sequence plus the transcriptional regulatory sequence (TRS) of sgRNA8 encoding the viral capsid protein N].
Immunoblotting
Virus-infected cells were resuspended in RIPA buffer (Sigma-Aldrich) supplemented with an EDTA-free protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and incubated on ice for 20 min to completely inactivate viruses. Cleared cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the proteins were transferred to the polyvinylidene difluoride membrane (GE Healthcare Life Sciences, Piscataway, NJ, USA). Following blocking with 5% bovine serum albumin, the blots were probed with an appropriate set of primary and secondary antibodies. Antibodies were obtained as follows: rabbit polyclonal anti-S (40069-T52; 1:1,000 dilution) and anti-N antibody (100211-RP02; 1:1,000 dilution) from Sino Biological Inc. (Beijing, China), and mouse monoclonal anti-α-tubulin antibody (DM1A; 1:5,000 dilution) from Calbiochem (La Jolla, CA, USA). Proteins were visualized using enhanced chemiluminescence.
Northern blotting
Total RNA extracted from the infected cells was denatured in a denaturing RNA loading buffer by incubation at 65°C for 5 min prior to gel electrophoresis on a 0.7% denaturing agarose gel. Following visualization of RNAs by ethidium bromide (EtBr) staining, the separated RNAs were transferred to a positively charged nylon membrane (Roche) via capillary action overnight and crosslinked by UV light. The membrane was then prehybridized using ULTRAhyb Ultrasensitive Hybridization Buffer (Thermo Fisher Scientific). DNA probe corresponding to the MERS-CoV genome ranging from nt 29,161–30,055 was radiolabeled with [α-32P]dCTP using the Rediprime II DNA labelling system (GE Healthcare Life Sciences). Hybridization of the DNA probe was carried out at 42°C in a hybridization buffer for 24 h. Membranes were washed and subjected to image analysis using an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare Life Sciences).
Fluorescence microscopy
Cells grown on a Lab-Tek 4-well chamber slide (Nunc, Roskilde, Denmark) were infected with MERS-CoV YKC-RFP at an MOI of 0.001. Two days later, cells were washed and fixed with 4% paraformaldehyde (Sigma-Aldrich). Fluorescence images were obtained using a confocal laser scanning microscope (Zeiss LSM 510 META, Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Prism Software Inc., La Jolla, CA, USA). Differences in means were analysed using a two-tailed, unpaired Student’s t-test. Unless otherwise specified, quantitative results were presented as mean ± standard deviation (SD) of at least three independent experiments. A P value of less than 0.05 was considered statistically significant.
Results
Construction of a full-length cDNA clone of a MERS-CoV Korean isolate
The full-length clone constructed in this study was generated using viral RNA obtained from the MERS-CoV KNIH002 strain (KOR/KNIH/002_05_2015: KT029139) [Citation8]. This strain belonging to the contemporary epidemic clade B was isolated from the spouse (Patient #2) of the index patient (Patient #1) during the 2015 MERS outbreak in South Korea. The virus was passaged thrice in Vero E6 cells for virus isolation and stock preparation, and the purified virus was further propagated in Huh7 cells to obtain viral RNA used for cDNA synthesis (A).
Initially, we prepared five overlapping cDNA fragments, namely M-CoV-B1, M-CoV-1, M-CoV-2, M-CoV-3, and M-CoV-4, which represent the nucleotides (nt) 1–811, 791–9,084, 9,065–18,784, 18,765–25,767, and 25,748–29,176 of MERS-CoV genome, respectively, by RT–PCR using sets of primers listed in . These fragments were cloned into a low copy number vector, pMW119, for sequencing analysis. For assembly of these cDNAs, one StuI site in the BAC plasmid was removed by site-directed mutagenesis. Similarly, one StuI site in the M-CoV-1 fragment (nt 791–9,084) was also removed by incorporating a G7622C substitution [resulting in no amino acid (aa) change], which also serves as a signature marker of the final infectious clone (B).
Sequencing of the resulting cloned cDNAs identified 9 nucleotide changes (one 2-nt deletion and 8 point mutations) compared with the genome sequence of KNIH002. To ensure that these were not artificially introduced during the cloning process, we additionally analysed 6 independent cDNA clones (1.5∼2.5 kb in size) covering these mutations (). Interestingly, we found a 2-nt deletion at nt 26,886–26,887 or nt 26,887–26,888 (due to the three consecutive T residues at nt 26,886–26,888, hereafter arbitrarily assigned to nt 26,886–26,887) in 2 out of 6 clones analysed. This deletion causes a frameshift that would produce, if any, a 22 aa-long peptide by introducing a premature stop codon. The G29097 T nt substitution at the nucleocapsid N-coding gene was also observed repeatedly albeit at a low frequency (1 out of 6 clones). These two changes were corrected in the cDNA clones according to the sequence of KNIH002. The remaining 8 point mutations (five silent mutations G19075A, T23303C, C24383 T, T25968A, and T26109C) and three nonsynonymous mutations [C6286 T, C11061 T, and T26109C, resulting in A2003V and P3595S substitutions in nonstructural protein 3 (Nsp3) and Nsp6 proteins encoded by ORF1a/b, and an M6 T substitution in ORF4b] were consistently found in all of the clones analysed. We thus incorporated these cell culture-adaptive mutations into the full-length clone.
Table 2. Summary of sequence analysis of the 4 cDNA clones used for construction of a full-length cDNA clone of KNIH002 (KT029139).
The sequence edited, overlapping cDNA fragments (M-CoV-1, M-CoV-2, M-CoV-3, and M-CoV-4) were sequentially assembled into a cassette plasmid pBAC-MERS-CoV/YKC-5′-3′ constructed as described in the Methods section. The assembled pBAC-MERS-CoV/YKC (Yonsei and KCDC) was sequenced to ensure that no additional mutations were introduced during the cloning steps. The full-length YKC clone had three amino acid changes (A2003V in Nsp3, P3595S in Nsp6, and M6 T in ORF4b) compared with its parental strain KNIH002.
Infectivity of the rescued recombinant MERS-CoV YKC
Using the full-length cDNA cloned in a BAC plasmid, P0 (passage #0) YKC virus was rescued by plating pBAC-MERS-CoV/YKC-transfected BHK-21 cells over Huh7 monolayers grown to confluence, as previously described [Citation20]. Infection of Huh7 with YKC P0 resulted in cell rounding and syncytia formation at 2 days post-infection (dpi), indicatives of CPE induced by MERS-CoV infection (A). Expression of viral capsid (N) and spike (S) proteins was verified by immunoblotting analysis of lysates of YKC P0-infected Huh7 cells (B). Furthermore, full-length viral genomic RNA (gRNA), as well as seven different sgRNAs, were detected by Northern blotting analysis of total RNA extracted from the infected cells (C). In addition, we verified viral sgRNA synthesis by detecting the TRS correctly fused to the N protein-coding gene through sequencing analysis of the 5′-end region of capsid protein (N)-coding sgRNA8 (D). Lastly, the G7622C genetic marker signifying YKC identity was confirmed by sequencing of the corresponding region that was RT–PCR amplified from viral RNA extracted from the YKC P0 (E).
Figure 2. Infectivity and replication competence of the recombinant MERS-CoV YKC rescued from the full-length cDNA clone of KNIH002. (A) The cytopathic effect induced by YKC P0 on Huh7 cells. (B) Immunoblotting analysis for the indicated viral structural proteins 2 days after infection of Huh7 cells with the P0 virus. α-tubulin was used as a loading control. (C) Northern blotting for detection of viral genome and subgenomic mRNAs at 2 dpi using a radiolabeled probe specific to ∼1 kb 3′-end region of the MERS-CoV genome. Visualization of 28S and 18S rRNA by ethidium bromide staining of total RNA separated by denaturing agarose gel electrophoresis (right panel). (D) Nucleotide sequence of the sgRNA encoding N protein (sgRNA8-N). The target gene was RT-PCR amplified using total RNA recovered from P0 YKC-infected Huh7 cells at 2 dpi. Shown in the enlarged chromatogram is the transcriptional regulatory sequence (TRS) flanked by the leader sequence and capsid protein (N)-coding gene. (E) The signature sequence (G7622C in ORF1a) of the infectious cDNA clone, verified by sequencing analysis of the corresponding region. (F) Vero E6 cells infected with MERS-CoV YKC at an MOI of 0.001 were subjected to immunoblotting analysis for the viral capsid protein N and the spike protein S (along with its two major cleaved forms). SFL, full-length S protein. Shown on the right of immunoblot is an illustration depicting proteolytic cleavage sites (S1/S2 and S2′) in S protein and its cleaved fragments, with their predicted molecular masses. SP, signal peptide; RBD, receptor-binding domain; RBM, receptor-binding motif; FP, fusion peptide; HR1 and HR2, heptad repeat 1 and 2; TM, transmembrane; CP, cytoplasmic tail. A single asterisk indicates a non-specific band, and a double asterisk indicates degraded forms of S protein. α-tubulin was used as a loading control.
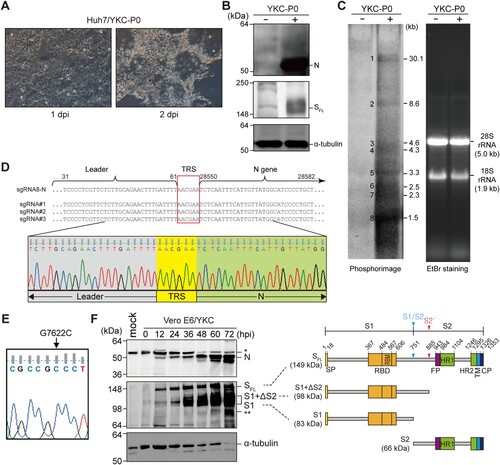
Figure 3. Analysis of the infectivity of the tdTomato RFP-expressing recombinant YKC, YKC-RFP. (A) A schematic illustration of the MERS-CoV YKC-RFP in which the ORF5 gene in YKC was replaced with tdTomato gene (top panel). Nucleotide sequence of the 5′-end region of the viral mRNA encoding the RFP (middle panel). TRS, transcriptional regulatory sequence. Fluorescence microscopic images of YKC-RFP (P2)-infected Vero E6 cells at 2 dpi (bottom panel). Scale bar, 100 µm. (B and C) Infectious virus titres and viral protein levels in Vero E6 cells infected with YKC (P1) or YKC-RFP (P1) at an MOI of 0.001. The results are the mean ± SD of two independent experiments with two replicates.
Moreover, the YKC P1 (passage #1 of YKC P0) propagated efficiently in Vero E6 cells. Viral structural proteins (N and S as well as its processed forms) were detected as early as 12 hpi and their expression levels increased gradually up to day 3 (F). Altogether, these data indicate that the recombinant virus (YKC strain) rescued from the established full-length clone of the Korean isolate KNIH002 strain was infectious.
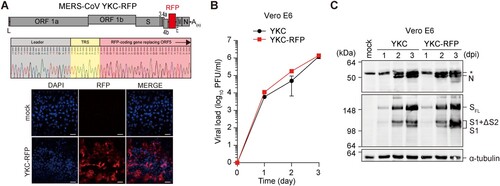
A recombinant YKC derivative expressing red fluorescent protein
Reporter-expressing infectious MERS-CoV is a valuable tool for antiviral drug discovery and for analysis of in vivo trafficking of MERS-CoV. We constructed a recombinant YKC derivative expressing the red fluorescent protein (RFP) tdTomato. Frequent occurrence of ORF5 deletion variants in KNIH002 () and previous studies [Citation16,Citation17] suggested the non-essential function of this accessory protein in viral propagation in vitro. We replaced the ORF5 with the tdTomato RFP gene to generate pBAC-MERS-CoV/YKC-RFP (A). The red fluorescence signal from tdTomato was detected at 2 dpi of Vero E6 cells with the recovered recombinant virus. Further, we confirmed the expression of sgRNA5-RFP transcripts bearing the TRS correctly fused to the RFP-coding gene by sequencing analysis of the 5′-end region of this mRNA.
The rescued, RFP-expressing virus (YKC-RFP; P1) displayed growth kinetics similar to that of YKC (P1) in Vero E6 cells (B). They expressed similar levels of N and S proteins during the course of infection (C), suggesting that deletion of ORF5 had little effect on either upstream (S) or downstream (N) sgRNA expression. Our results were in part consistent with previous studies showing that the ORF5 is dispensable for viral propagation, as demonstrated with EMC strain-based infectious cDNA clones lacking the ORF5 gene [Citation16,Citation17]. However, the recombinant virus did not show retarded growth compared with the parental virus YKC, which was in contrast to the results of a previous study in which replacement of ORF5 with the RFP gene was shown to be associated with a substantial reduction in replication. This discrepancy might be due to difference in reporter gene expression levels or was caused by genetic variaions between EMC and YKC strains.
Of note, the ORF5-deleted recombinant virus used in the previous study was constructed in the backbone of an infectious cDNA clone of EMC strain (P9) [Citation17].
Comparison of growth kinetics of the clade B MERS-CoV Korean strain and the clade A EMC strain
The nucleotide identity between the prototype MERS-CoV EMC strain (clade A) and the Korean isolate KNIH002 (clade B) was as high as 99.57% (29,978 nt identical among 30,108 nt in total). Nevertheless, there were 44 amino acid changes derived from 130 nucleotide differences between these two strains (A). As a first step to unravel the molecular mechanisms behind pathophysiological differences between these two strains, we compared the growth kinetics of EMC (unknown passage number) and KNIH002 (P5) along with its derivative YKC (P1) in Vero E6 cells. As shown in B, among them, the EMC strain yielded a significantly higher infectious virus titre than the Korean isolate KNIH002 and its derivative clone YKC. EMC reached the plateau titre of 4 ×107 PFU/ml at 48 hpi while KNIH002 and YKC strains grew slowly, reaching a peak titre of approximately 106 PFU/ml at 72 hpi. Interestingly, the growth of the YKC strain was further attenuated compared to its parental strain. The KNIH002 strain yielded approximately 0.3-log10 higher viral titres than YKC at 72 hpi and more substantially elevated titres in the earlier period of infection.
Figure 4. Growth attenuation features of KNIH002 and YKC. (A) The 44 amino acid differences within various ORFs between EMC and KNIH002 strains. (B–E) Infectious virus titre was determined at the indicated time points following infection of Vero E6 cells with MERS-CoV EMC, KNIH002, or YKC at an MOI of 0.001. The results are the mean ± SD of two independent experiments with two replicates (B). Immunoblotting for N and S proteins at 24 and 60 hpi. (C). Copy number of N protein-coding viral sgRNA (sgRNA8-N) determined by RT-qPCR at 60 hpi (D). The average and range of two technical replicates is shown. Plaque size was measured at 4 dpi of Vero E6 cells (EMC, n = 23, KNIH002, n = 23; YKC, n = 18). Shown under the scatter plots are representative plaque morphologies with mean diameters ± SD (mm) of plaques (E). The asterisk in (C) indicates a nonspecific band. In (B and E), red and blue asterisks indicate the comparison of the EMC strain with KNIH002 and with YKC strains respectively; black asterisks indicate the comparison between KNIH002 and YKC strains. **P < 0.01; ***P < 0.001; ****P < 0.0001.
This growth attenuation feature of YKC was more straightforwardly evidenced by reduced expressions of N and S proteins at 60 hpi compared to the EMC strain, and their expression levels were further decreased in cells infected with YKC compared to KNIH002 (C). Moreover, the highest sgRNA8-N titre of the EMC strain was > 1-log10 higher than that of KNIH002 or YKC (D). In consistency with these overall attenuated growth characteristics of KNIH002 and YKC, EMC infection formed the largest plaques, with an area of 16.9 ± 4.3 mm2 at 3 dpi (E). Further, the plaque size of YKC was significantly reduced (approximately 60%) compared with that of KNIH002 (P = 0.0051 by two-tailed, unpaired t-test).
Rapid emergence of diverse ORF5 variants during serial passages of MERS-CoV
During sequencing analysis of the cDNAs we prepared for generation of a full-length cDNA clone of KNIH002 strain, we found various ORF5 deletion variants in the P6 KNIH002 stock (A and see details in ). Taking advantage of the full-length clone of KNIH002, we addressed the question of how rapidly and frequently ORF5 variants can emerge in vitro in the absence of the selection pressure of host immune responses. After a single-step growth following pBAC-MERS-CoV/YKC transfection into BHK21 and subsequent co-culture with Huh7 cells, we found only a single point mutation at nt 27,020 (resulting in a C61G aa substitution) in the ORF5 of YKC (P0) with a frequency of 3 out of 6 clones (B).
Figure 5. Genetic instability of the ORF5 of MERS-CoV. (A) Various ORF5 deletion variants in KNIH002 (P6). (B) Identification and localization of mutations emerged following three consecutive passages (P3) of the YKC P0 rescued from the infectious cDNA clone of the KNIH002 strain. A total of 6 clones were analysed for the detection of variants. del, deletion; ins, insertion.
To our surprise, we found diverse ORF5 variants accumulating after three consecutive passages of YKC P0 in Vero E6 cells, which do not produce type I interferon (IFN) due to a genetic deletion of type I IFN genes [Citation24] and low endogenous level of IFN regulatory factor 3 (IRF-3), a transcription factor required for IFN induction [Citation25]. Of six independent ORF5 cDNA clones, we could detect only one clone remaining unchanged (P3_#1). The single point mutation causing a C61G substitution, which was detected in P0, was persistently carried over in P3. This nonsynonymous mutation was observed with the same frequency of 3 out of 6 in variants bearing either a deletion or an insertion mutation. Deletions of various lengths were detected in the TRS [7-nt (nt 26,829–26,835) deletion in P3_#3; expected to produce no ORF5 sgRNA since sgRNA synthesis would be abolished due to the absence of TRS] and in coding sequence [16-nt (nt 26,857–26,872) deletion in P3_#4; 6-nt (nt 27,186–27,191) deletion in P3_#5]. An insertional mutation harbouring an 11-nt-long sequence complementary to the Spike protein-coding gene (nt 23,554–23,564) was observed in one variant (P3_#6).
Our results show how rapidly viral quasispecies with diverse mutations in ORF5 could be generated due to the intrinsic error-prone nature of viral RNA polymerase and possibly through imprecise RNA recombination events during serial passages of MERS-CoV.
Discussion
In this study, we established a BAC-based reverse genetics system for a Korean isolate of MERS-CoV, KNIH002 strain. The first full-length infectious clone YKC for the clade B isolate would be useful for the investigation of pathogenesis and virus-host interactions through a comparative study with infectious clones of the clade A EMC strain, which displayed pathophysiologically distinctive features compared with MERS-CoV isolates from Korea [Citation10,Citation11,Citation26]. During the analysis of the infectivity of recombinant virus rescued from the full-length cDNA clone, we found that growth attenuation in cell culture was a distinctive phenotypic difference between KNIH002 (as well as YKC) and EMC strains. The retarded growth of KNIH002 must be genetically linked to the 44 aa changes in various regions of the viral genome, which individually or together would affect either virus entry, replication competence, and/or ability to evade innate immune responses.
The receptor-binding domain (RBD) of S protein was shown to have multiple aa sequence differences between EMC strain and various MERS-CoV strains isolated from patients during the outbreak in South Korea [Citation27]. In the RBD, there was only one aa difference V530L between EMC and KNIH002 strains, while two other changes, S137R and Q1020R were localized at sites other than the RBD of S protein (aa 367–606). Importantly, these mutations did not affect the processing of S protein (C). KNIH002 and YKC carrying these mutations produced significantly reduced levels of infectious viruses and viral structural proteins (S and N proteins) at early time points of infection (12 and 24 hpi) (B and C). These findings raised the possibility that the mutations in the S protein of KNIH002 and YKC, particularly the ones introducing the positively charged Arg residue at the sites outside the RBD, might influence viral entry possibly through altering its affinity toward the viral receptor dipeptidyl peptidase 4 (DPP4). A previous study showed that the two mutations D510G and I529 T in the RBD led to weaker binding of S protein to DPP4 and consequently hindered virus entry when an entry assay was carried out using pseudotyped viruses [Citation12]. These unique mutations detected in some, but not all, of the Korean isolates were not present in KNIH002 and YKC.
The viral Nsps encoded by the ORF1a/b of MERS-CoV are considered to be essential for viral genome replication [Citation28]. Among them, the Nsp3 had the greatest number of mutations resulting in 14 aa differences between EMC and KNIH002 strains. By contrast, the Nsp12 viral RNA-dependent RNA polymerase essential for viral replication [Citation29] was well conserved in both strains. These biased mutation profiles suggested that the mutations were unlikely introduced at random positions during synthesis and amplification of viral cDNAs. Besides its papain-like protease activity, Nsp3 plays a key role in viral replication by acting as a scaffold protein interacting with other viral Nsps and/or host proteins and acts as an antagonist of IFN responses [Citation30]. The finding of 14 aa substitutions scattered across different domains of this multifunctional protein opens gates for further research regarding the influence of these genetic variations on viral growth.
The coronavirus accessory ORF proteins, which are least conserved in the CoV family, appear to be dispensable for replication while their deletions resulted in attenuation of MERS-CoV growth in vitro and in mice [Citation21,Citation31]. Although a fewer number of aa differences was found in the ORF3 accessory protein, the percentage of differences normalized to protein molecular mass was higher in ORF3 than in Nsp3. Its function is still unknown. As such, the impact of mutations on ORF3 remains to be characterized. The two other accessory proteins, ORF4b and ORF5 are known to act as IFN antagonists [Citation32]. Further the structural protein M (membrane protein), which is proposed to be involved in virus assembly through its interaction with the viral capsid protein N, is also known to antagonize IFN action [Citation32]. Thus, dysregulation of the function of these IFN antagonists by mutations would influence viral growth. The two substitutions V69I and F123I in the M protein may account for the difference in growth rate between EMC and KNIH002 strains. Previous studies with EMC ORF4b revealed that it prevents NF-κB-mediated innate immune response [Citation33] and RNase L activation induced by type I IFN [Citation34]. It would be of interest to investigate if these functions of ORF4b protein are influenced by mutations found in KNIH002 and YKC strains. Taken together, variations in Nsps, accessory ORF proteins, and even in the structural proteins (S, M, and N) would play together in establishing phenotypic differences between EMC and KNIH002 strains, for example, not only in growth but also in terms of immune evasion capacity, transmissibility, and pathogenicity.
Further, it is worth pointing out that the growth of YKC was further attenuated in comparison to its parental strain by acquiring three additional aa changes (A2003V in Nsp3, P3595S in Nsp6, and M6 T in ORF4b) introduced during consecutive passages of KNIH002. Since S protein sequence was conserved between KNIH002 and YKC strains, it is plausible to speculate that the aa changes in Nsps, individually or together with the M6 T substitution in the ORF4b, account for the reduced growth of YKC. Further, the additional five nucleotide changes, which lead to synonymous mutations (G19075A, T23303C, C24383 T, T25968A, and T26109C), may also contribute to the slower growth of YKC, as these nucleotide changes even within the ORFs might alter RNA conformations to dysregulate viral gene expression and/or replication.
Sequencing analysis of clinical samples, which were not cultivated in vitro, identified various deletions variants. For instance, various mutants detected in nasopharyngeal swab specimens collected from a Korean patient travelling to China in late May 2015 had deletions at the junction of ORF5 and E genes, resulting in the fusion between the ORF5 and E proteins or a deletion at the C-terminus of ORF5 [Citation35]. An earlier study also identified deletions in ORF3 and ORF4a genes in clinical samples collected during the outbreak in Riyadh in 2015 [Citation36]. The effect of these deletions in accessory ORFs of MERS-CoV and the molecular mechanism of biogenesis of these defective accessory ORF genes are unknown. Recently, more diverse deletion variants were detected in the clade B MERS-CoV isolates collected from patients in Saudi Arabia in 2015 [Citation37]. The authors found deletions in the 5′-UTR, Nsp2 (in ORF1a), and ORF3. While deletions in the 5′-UTR and ORF1a resulted in reduced viral RNA titres in culture media, the effect of deletion in ORF3 has not yet been addressed. Our observations of ORF5 deletion variants are reminiscent of these previous findings. We found that the ORF5 of MERS-CoV undergoes rapid mutations including even insertion and deletion (). During sequence analysis of KNIH002 cDNA clones, we repeatedly detected an ORF5 variant with a 2-nt-deletion that would produce a prematurely terminated 22-aa long peptide, although it was not a predominant one ().
More importantly, following passages of the recombinant virus rescued from the YKC clone, we could detect diverse ORF5 variants including truncated ORF5 variants, which can produce 107-aa long and 222-aa long ORF5 proteins through premature termination caused by internal insertion and deletion, respectively. Notably, an ORF5-deleted mutant virus generated by engineering an infectious clone of EMC strain remained infectious [Citation16,Citation17]. Our results also showed that the replacement of ORF5 with RFP had little effect on the growth of YKC-RFP. Whilst these results support the non-essential role of ORF5 in viral propagation in vitro, it is worth investigating whether there are any functional roles of deletion variants of ORF5 protein. Further studies are required to gain some insights into how these highly metastable genes of MERS-CoV have evolved and will change in non-human networks, and play roles in the adaptation of MERS-CoV in different hosts. Viral pathogenesis and fitness in host cells are likely to be influenced by genetic variations in viral genome and consequently the quasispecies dynamics of MERS-CoV. As exemplified by the rapid emergence of ORF5 variants following three consecutive passages of YKC P0 viral stock, quasispecies are likely to be present in viral stocks as clinical specimens had to be passaged multiple times for virus purification by plaque-forming assay or a limited-dilution procedure and for amplification of purified virus. Our results call for caution in the uses of viral stocks harbouring unidentified variants in pathogenesis studies.
Our study had several limitations because original virus stocks of EMC and KNIH002 strains used for comparative growth kinetic study have been passaged multiple times in vitro following isolation from patients and sequencing analysis. The reference sequences of KNIH002 and EMC are for the P3 and P6 stocks, respectively. For growth kinetic comparison, we used KNIH002 P5 and EMC with an unknown passage number (likely multiple more passages after sequencing analysis using a P6 stock) [Citation2]. Thus, the reference sequences of these viruses and the sequences of the major population of the viral stocks used in this study are likely to have variations. Additionally, the lack of viral sequences for clinical samples limits our ability to differentiate the contribution of culture-adapted mutations in conferring phenotypic differences between the EMC and KNIH002 strains. In this regard, the infectious cDNA clones now available for both clade A and clade B strains of MERS-CoV will enable us to investigate systemically how viral pathogenesis and transmissibility of MERS-CoV are influenced by the genetic drifts we observed between the two representative MERS-CoV strains.
One of the practical implications of the infectious clone established in this study is to use it as a backbone for the development of live attenuated vaccines by taking advantage of the growth attenuated feature of the recombinant virus YKC. This phenotype is supposed to be inherited relatively stably as it is genetically connected to 130 nucleotide changes leading to 44 amino acid substitutions. An earlier proof-of-concept study indeed demonstrated the potential use of a growth attenuated Nsp16 mutant carrying a single aa change (D130A) in the active site of viral 2′O-methyltransferase [Citation22]. Additionally, the recombinant MERS-CoV expressing RFP (YKC-RFP) will facilitate high-throughput screening of antiviral drugs and in vivo tracking of MERS-CoV through imaging analysis.
Conclusion
In summary, our study demonstrates a remarkable difference in growth kinetics between two representative MERS-CoV strains isolated during the outbreaks in Saudi Arabia in 2002 and South Korea in 2015. We provide experimental evidence of the genetic instability of MERS-CoV ORF5 using the infectious cDNA clone we established for the clade B MERS-CoV Korean isolate KNIH002. As exemplified by ORF5 variants, viral quasispecies, which would be unavoidably produced during serial passages of virus samples, should be taken into considerations when investigating the pathogenesis and evaluating the efficacy of control measures such as neutralizing antibodies and antiviral agents against MERS-CoV. Use of a low-passage virus rescued from infectious cDNA clones now available for MERS-CoV isolates in clades A and B is the option to avoid the potential interference from viral quasispecies produced due to low fidelity of MERS-CoV Nsp12 RdRp and by aberrant RNA recombination events.
Acknowledgements
We thank Bart Haagmans (Department of Viroscience, Erasmus Medical Center, Rotterdam, the Netherlands) and Luis Enjuanes (Campus Universidad Autónoma, Cantoblanco, Madrid, Spain) for reagents.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis. 2014;59(9):1225–1233.
- Zaki AM, van Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820.
- Hakawi A, Rose EB, Biggs HM, et al. Middle East respiratory syndrome coronavirus, Saudi Arabia, 2017-2018. Emerg Infect Dis. 2019;25(11):2149–2151.
- Payne DC, Biggs HM, Al-Abdallat MM, et al. Multihospital outbreak of a Middle East respiratory syndrome coronavirus deletion variant, Jordan: a molecular, serologic, and epidemiologic investigation. Open Forum Infect Dis. 2018;5(5):ofy095.
- Donnelly CA, Malik MR, Elkholy A, et al. Worldwide reduction in MERS cases and deaths since 2016. Emerg Infect Dis. 2019;25(9):1758–1760.
- Korea Centers for Disease C, Prevention. Middle East respiratory syndrome coronavirus outbreak in the Republic of Korea, 2015. Osong Public Health Res Perspect. 2015;6(4):269–278.
- Kim SW, Park JW, Jung HD, et al. Risk factors for transmission of Middle East respiratory syndrome coronavirus infection during the 2015 outbreak in South Korea. Clin Infect Dis. 2017;64(4):551–557.
- Kim YJ, Cho YJ, Kim DW, et al. Complete genome sequence of Middle East respiratory syndrome coronavirus KOR/KNIH/002_05_2015, isolated in South Korea. Genome Announc. 2015;3(4):e00787-00715.
- Kim KH, Tandi TE, Choi JW, et al. Middle East respiratory syndrome coronavirus (MERS-CoV) outbreak in South Korea, 2015: epidemiology, characteristics and public health implications. J Hosp Infect. 2017;95(2):207–213.
- Al-Tawfiq JA, Assiri A, Memish ZA. Middle East respiratory syndrome novel corona (MERS-CoV) infection epidemiology and outcome update. Saudi Med J. 2013;34(10):991–994.
- Breban R, Riou J, Fontanet A. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: estimation of pandemic risk. Lancet. 2013;382(9893):694–699.
- Kim Y, Cheon S, Min CK, et al. Spread of mutant Middle East respiratory syndrome coronavirus with reduced affinity to human CD26 during the South Korean outbreak. MBio. 2016;7(2):e00019.
- Kleine-Weber H, Elzayat MT, Wang LS, et al. Mutations in the spike protein of Middle East respiratory syndrome coronavirus transmitted in Korea increase resistance to antibody-mediated neutralization. J Virol. 2019;93(2):e01381–18.
- Kim YS, Aigerim A, Park U, et al. Sequential emergence and wide spread of neutralization escape Middle East respiratory syndrome coronavirus mutants, South Korea, 2015. Emerg Infect Dis. 2019;25(6):1161–1168.
- Almazan F, Sola I, Zuniga S, et al. Coronavirus reverse genetic systems: infectious clones and replicons. Virus Res. 2014;189:262–270.
- Almazan F, DeDiego ML, Sola I, et al. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. MBio. 2013;4(5):e00650–13.
- Scobey T, Yount BL, Sims AC, et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc Natl Acad Sci USA. 2013;110(40):16157–16162.
- Nikiforuk AM, Leung A, Cook BWM, et al. Rapid one-step construction of a Middle East respiratory syndrome (MERS-CoV) infectious clone system by homologous recombination. J Virol Methods. 2016;236:178–183.
- Lau SK, Wernery R, Wong EY, et al. Polyphyletic origin of MERS coronaviruses and isolation of a novel clade A strain from dromedary camels in the United Arab Emirates. Emerg Microbes Infect. 2016;5(12):e128.
- Almazan F, Marquez-Jurado S, Nogales A, et al. Engineering infectious cDNAs of coronavirus as bacterial artificial chromosomes. Methods Mol Biol. 2015;1282:135–152.
- Ahn DG, Lee W, Choi JK, et al. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res. 2011;91(1):1–10.
- Menachery VD, Gralinski LE, Mitchell HD, et al. Middle East respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere. 2017;2(6):e00346–00317.
- Zhou J, Li C, Zhao G, et al. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci Adv. 2017;3(11):eaao4966.
- Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol. 1979;43(1):247–252.
- Chew T, Noyce R, Collins SE, et al. Characterization of the interferon regulatory factor 3-mediated antiviral response in a cell line deficient for IFN production. Mol Immunol. 2009;46(3):393–399.
- Sabir JS, Lam TT, Ahmed MM, et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science. 2016;351(6268):81–84.
- Kim DW, Kim YJ, Park SH, et al. Variations in spike glycoprotein gene of MERS-CoV, South Korea, 2015. Emerg Infect Dis; 2016;22(1):100–104.
- Wang Y, Grunewald M, Perlman S. Coronaviruses: An updated overview of their replication and pathogenesis. Methods Mol Biol. 2020;2203:1–29.
- Ahn DG, Choi JK, Taylor DR, et al. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch Virol. 2012;157(11):2095–2104.
- Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58–74.
- Menachery VD, Mitchell HD, Cockrell AS, et al. MERS-CoV accessory ORFs play key role for infection and pathogenesis. MBio. 2017;8(4):e00665-00617.
- Yang Y, Zhang L, Geng H, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4(12):951–961.
- Canton J, Fehr AR, Fernandez-Delgado R, et al. MERS-CoV 4b protein interferes with the NF-kappaB-dependent innate immune response during infection. PLoS Pathog. 2018;14(1):e1006838.
- Thornbrough JM, Jha BK, Yount B, et al. Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. mBio. 2016;7(2):e00258.
- Xie Q, Cao Y, Su J, et al. Two deletion variants of Middle East respiratory syndrome coronavirus found in a patient with characteristic symptoms. Arch Virol. 2017;162(8):2445–2449.
- Lamers MM, Raj VS, Shafei M, et al. Deletion variants of Middle East respiratory syndrome coronavirus from humans, Jordan, 2015. Emerg Infect Dis; 2016;22(4):716–719.
- Tamin A, Queen K, Paden CR, et al. Isolation and growth characterization of novel full length and deletion mutant human MERS-CoV strains from clinical specimens collected during 2015. J Gen Virol. 2019;100(11):1523–1529.