
ABSTRACT
ST59 is the predominant pathotype of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) in China. As a variant of ST59, there is relatively little known about the detailed information of ST338. To address this issue, here, we described thirteen ST338 CA-MRSA strains isolated from severe bloodstream infection cases, and focused on their epidemiology, genetic features and virulence potential. Phylogenetic analysis showed the earliest isolated strain of this study is likely a predecessor of recent ST338 lineage (after year of 2014). Furthermore, the phylogenetic reconstruction and time estimation suggested that ST338 evolved from ST59 in 1991. Notably, the carrying patten of virulence factors of all ST338 strains were similar, and the genomic islands νSaα, νSaγ and SaPI and the core virulence factors like hla and psm were detected in ST338 isolates. However, all ST338 isolates lacked some adhesion factors such as clfA, clfB, eap, cna and icaD. Additionally, among these ST338 strains, one PVL-negative ST338 isolate was detected. Experiment on mice nose and human alveolar epithelial cell showed that the nasal colonization ability of ST338 was weaker than that of CA-MRSA MW2. In a mouse bloodstream infection model and skin infection model, PVL+ and PVL− strains had the similar virulence, which was dependent on upregulation of toxin genes rather than the presence of mobile genetic elements such as ΦSa2 carrying PVL. Our findings provide important insight into the epidemiology and pathogenicity of the novel and highly virulent ST338-SCCmec Vb clone.
Introduction
Staphylococcus aureus is a common opportunistic pathogen that usually colonizes human skin and nasal mucosa and causes a variety of infections ranging from mild skin lesions to severe diseases such as endocarditis, osteomyelitis, pneumonia, and even life-threatening septic shock [Citation1–3].
S. aureus can acquire resistance in various ways, which has led to the emergence of community-associated methicillin-resistant S. aureus (CA-MRSA), a major cause of infections in hospitals worldwide [Citation4], and the most common cause of skin and soft tissue infections (SSTIs) in the United States [Citation5–7]. Unlike healthcare-associated (HA)-MRSA, which is mostly isolated from older patients with underlying health problems and risk factors [Citation8], CA-MRSA tends to infect healthy younger individuals and is more virulent, potentially causing severe pneumonia and fatal sepsis [Citation9]. Some CA-MRSA clones have a high rate of dissemination within the general population.
CA-MRSA harbours type IV or V staphylococcal chromosomal cassette mec (SCCmec) elements that are smaller than the type II or III SCCmec elements of HA-MRSA [Citation10]. Unlike HA-MRSA, CA-MRSA strains are sensitive to many non-β-lactam antibiotics and carry Panton–Valentine leukocidin (PVL) genes [Citation11]. CA-MRSA strains were once limited to populations outside health care settings, which was the basis for their differentiation from HA-MRSA. However, this distinction has been obscured in recent years and the prevalence of CA-MRSA strains has increased; thus, the epidemiology of MRSA has changed with the emergence of CA-MRSA strains in healthcare settings. The epidemiologic success of CA-MRSA is attributed to a combination of low fitness cost of antibiotic resistance and high virulence [Citation12–14]. However, most studies on CA-MRSA in China have focused on the ST59 clone, and there is little information on other clones.
ST59, the predominant CA-MRSA clone in China, has 2 variants: a PVL+ Taiwan clone (ST59-SCCmec V) and PVL− Asia-Pacific clone (ST59-SCCmec IV) [Citation15–17]. Studies conducted in Taiwan have reported that ST59-SCCmec V has a greater capacity to lyse neutrophils and causes more severe infections than ST59-SCCmec IV, although the latter more efficiently colonizes nasal mucosa [Citation18, Citation19]. ST338 is a variant of ST59, with a single-nucleotide mutation in the housekeeping gene gmk. The virulent ST59-SCCmec Vb clone recently emerged in Taiwan and mainland China [Citation20, Citation21], and its virulence determinants have been investigated [Citation22], but detailed information on ST338-SCCmec Vb is lacking.
The Blood Bacterial Resistance Investigation Collaborative System (BRICS) is a national surveillance programme that collects pathogens isolated from patients with bloodstream infection to monitor epidemics of bacteria in China [Citation23–25]. From January 2014 to December 2019, we isolated 927 MRSA strains from various provinces and cities in China; 13 of these were ST338-SCCmec Vb (SA01–SA13) classified as CA-MRSA according to the Centers for Disease Control and Prevention definition and caused severe bloodstream infections. In this study, we report 13 cases of severe septicaemia caused by SA01–SA13 and describe the epidemiology, genetic features, and virulence of the isolates. Among these CA-MRSA isolates, one PVL-negative ST338 isolate was detected. Thus, to identify the key elements in the evolution of virulence in CA-MRSA, we subsequently analysed the virulence factors in CA-MRSA ST338-SCCmec Vb.
Materials and methods
Bacterial strains and CA-MRSA definition
ST338 isolates were obtained from BRICS. CA-MRSA was defined as a strain isolated within 48 h from outpatients or patients admitted to the hospital who had no history of inpatient contact with nursing homes, shelters, or other health institutions within the previous 12 months; and had no indwelling catheter or artificial medical device.
Antimicrobial susceptibility testing
Antimicrobial susceptibility was assessed using the VITEK 2/VITEK 2 XL system (BioMérieux, Marcy l’Etoile, France). The minimum inhibitory concentrations (MICs) of 17 antimicrobial agents including ciprofloxacin, nitrofurantoin, levofloxacin, penicillin G, sulfamethoxazole and trimethoprim, teicoplanin, linezolid, tigecycline, quinupristin/dalfopristin, gentamicin, oxacillin, rifampin, tetracycline, vancomycin, clindamycin, moxifloxacin, and erythromycin were determined.
Whole-genome sequencing (WGS) and genomic analysis
Genomic DNA of ST338 and ST59 isolates was extracted using the Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech, Shanghai, China) and sequenced using the HiSeq X 10-PE150 platform (Illumina, San Diego, CA, USA). The quality of raw reads was verified using FastQC 0.11.7 software [Citation26] and the reads were trimmed using Trimmomatic v0.34 (Phred quality score >20) [Citation27]. Clean reads were assembled using SPAdes v3.13 [Citation28] with default settings. The assembly was filtered and contigs larger than 200 bp were retained. Multilocus sequence typing of S. aureus was performed by searching 7 housekeeping genes against the National Center for Biotechnology Information (NCBI) PubMLST database (https://github.com/tseemann/mlst). SCCmec typing was performed using SCCmecFinder (https://cge.cbs.dtu.dk/services/SCCmecFinder/). The sequences of 30 ST338 and 30 ST59 MRSA-SCCmec Vb strains from China and surrounding geographic regions were downloaded from GenBank for comparisons.
Phylogenetic analysis
We used Snippy v4.6.0 (https://github.com/tseemann/snippy) to identify single-nucleotide polymorphisms (SNPs) in the ST338 and ST59 strains, with M013 (GenBank accession no. CP003166) – a PVL+ CA-MRSA ST59 SCCmec Vb clone isolated in Taiwan [Citation29] – as a reference. Recombined regions were detected using Gubbins v2.4.1 [Citation30], and the output from Gubbins was used as the input for BactDating v1.0 [Citation31] to perform phylogenetic dating based on a Bayesian approach. The Markov chain Monte Carlo chain lengths were run for 100 million cycles to convergence; the effective sample size of the inferred parameters α, μ, and σ was >200. A minimum spanning tree (MST) based on SNP data from the 13 ST338 SCCmec Vb isolates in this study was constructed using CiteSpace [Citation32].
Identification of virulence genes and detection of S. aureus pathogenicity islands (SaPIs)
Sequences of S. aureus MW2 (ST1; GenBank accession no. BA000033), USA300 (ST8; GenBank assembly accession GCA_000013465.1), rh10 (ST59-SCCmec Vb from mainland China), and M013 (ST59-SCCmec Vb from Taiwan; GenBank accession no. CP003166) were used for comparative analyses of virulence. Genome sequences were annotated using the RAST server (https://rast.nmpdr.org/rast.cgi). Virulence factors were detected using the Virulence Factors Database analyser (http://www.mgc.ac.cn/VFs/). A genomic island map was generated using the genoplotR package of R software [Citation32].
Mouse nasal colonization model
Animal experiments were carried out in accordance with the guidelines for the Care and Use of Laboratory Animals of the Chinese Association for Laboratory Animal Sciences. The mouse nasal colonization model was established using 6-week-old female BALB/C mice purchased from the Animal Laboratory Center of the First Affiliated Hospital of Zhejiang University School of Medicine. A drop of phosphate-buffered saline (PBS; 20 μl) containing 1×108 S. aureus cells was introduced into the nasal passage of the mice (n=5 per S. aureus strain). After 3 days, the mice were sacrificed and the nose was excised and homogenized. Total S. aureus counts were determined by plating 200 µl of diluted nose tissue cell suspension on tryptic soy agar (TSA).
Adhesion of S. aureus to human alveolar epithelial cells
The adhesion assay was performed as previously described [Citation33]. Briefly, strains were grown for 6 h at 37°C and then washed 3 times with sterile PBS. A549 human alveolar epithelial cells and bacterial cells were coincubated at 37°C and 5% CO2 in a 6-well plate. After 3 h, dissociated bacterial cells were washed and 0.1% deoxysodium cholate solution was used to lyse the A549 cells.
Bacteraemia caused by MRSA in a mouse sepsis model
Female BALB/C mice (6 weeks old) were used to establish a sepsis model. Each mouse was housed in the laboratory for 10 days prior to the experiment. S. aureus strains were grown for 9 h and cells were harvested by centrifugation and washed 3 times with PBS; 1×108 cells were resuspended in 50 μl sterile PBS and injected into mice (n=10 per strain) via the caudal vein. Negative control mice were injected with 50 μl sterile PBS. The physical condition of inoculated mice was monitored and recorded daily. Mice were sacrificed as soon as they were unable to eat or drink. When the first mouse infected with ST338 died, all remaining mice in the group were sacrificed. At 7 days post infection, all surviving mice were sacrificed. The lungs were dissected, fixed in 10% formalin, and embedded in paraffin. Sections were cut and stained with haematoxylin and eosin. The left kidney was removed and homogenized in 1 ml PBS, and 200 μl of homogenized tissue was diluted and plated on TSA for S. aureus cell counts.
Quantitative reverse-transcription (qRT)-PCR
RNA was extracted from S. aureus strains using the RNeasy Purification Plus Kit (Qiagen, Hilden, Germany). Purified RNA was reverse transcribed to cDNA using SYBR Green Premix (Takara Bio, Otsu, Japan). Expression of hla, psmα, agrA, and RNAIII genes in ST338 was detected by qRT-PCR and quantified with the 2−ΔΔCt method. USA300 was used as a reference strain (relative expression=1) because of its high endogenous expression of these genes [Citation34, Citation35].
Red blood cell (RBC) lysis assay
Haemolytic capacity was evaluated as previously described [Citation36]. Briefly, S. aureus strains were grown for 9 h and centrifuged. The supernatant was incubated with rabbit RBCs for 3 h at 37°C. Purified water and rabbit RBCs resuspended in NaCl solution served as positive and negative controls, respectively.
Cytotoxicity assay
Neutrophils were extracted from the blood of healthy human volunteers using the Human Neutrophil Isolation Solution Kit (Sangon Biotech) and stored in cell-preserving solution. S. aureus strains were grown overnight at 37°C. After 1:200 dilution in PBS, 200 μl of the culture and neutrophils were coincubated in a 96-well microplate for 3 h at 37°C. Neutrophils incubated in 0.1% Triton-X100, TS broth, and cell-preserving solution served as positive, negative, and blank controls, respectively.
Statistical analysis
Assays were performed three times. The q test was used to evaluate differences in skin lesions between mice, and the Wilcoxon rank-sum test was used to compare colony-forming unit (CFU) counts. Survival rates of mice infected with S. aureus were analysed with the Kaplan–Meier method. qRT-PCR data were analysed with the unpaired t test. Results of the haemolysis test and cytotoxicity assay were analysed with the chi-squared test. Data analyses were performed with Prism v7.0 software (GraphPad, San Diego, CA, USA). P<0.05 was considered significant.
Results
Brief case reports
Of the 927 MRSA isolated from blood samples in China over a 6-year period, 13 ST338-SCCmec Vb isolates (ST338-SCCmec Vb; SA01–SA13) were obtained from severe CA-MRSA infection cases at different hospitals [(a)]. The mean age of patients was 32 years old. Nine cases were primary SSTIs; 7 patients ultimately died. The clinical information of these patients is presented in .
Figure 1. Relationships among ST338 isolates in this study. (a). Geographic distribution of clinical cases of ST338 infection in China from 2014 to 2019. Different colours represent areas where the strains were isolated. (b). MST of ST338 isolates. The genome sequences of 13 ST338 isolates were aligned. Each circle represents a ST338 strain, and each colour represents a different geographic region. Numbers on the connecting lines indicate the number of single nucleotide polymorphisms between two strains.
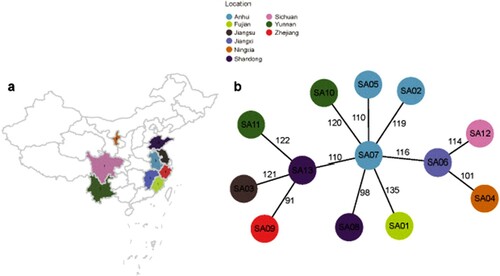
Table 1. Clinical reports of ST338 community-associated methicillin-resistant Staphylococcus aureus cases.
Characteristics of ST338 isolates
The 13 MRSA isolates were identified as ST338 SCCmec Vb (5C2&5) CA-MRSA strains (). All isolates were resistant to erythromycin, clindamycin, and oxacillin and had a methicillin MIC of 4 μg/ml, indicating very low methicillin resistance. The isolates were susceptible to trimethoprim, vancomycin, rifampin, levofloxacin, moxifloxacin, gentamicin, teicoplanin, amikacin, tigecycline, daptomycin, and linezolid.
Table 2. Genotypes and susceptibilities to 17 antibiotics in thirteen clinical MRSA isolates of ST338 in China.
Relationships among ST338 isolates in this study
To determine whether the CA-MRSA ST338 strains causing severe bloodstream infection are an epidemiologic threat, we screened 927 MRSA strains isolated from 17 provinces in China from 2014 to 2019. With the exception of Ningxia Province, all strains emerged in geographically close provinces [(a)], suggesting clonal transmission of ST338-SCCmec Vb. The MST revealed that all ST338 isolates were closely related (<150 SNPs). Strain SA07 [middle circle in (b)] was the earliest isolate identified in Anhui Province in 2014, while other strains were isolated after 2014, implying that SA07 is the ancestor of the other ST338-SCCmec Vb clones and has the potential to evolve by adapting to different environments through the acquisition of novel mutations.
Phylogenetic construction of ST338 SCCmec Vb clonotype MRSA and time estimation
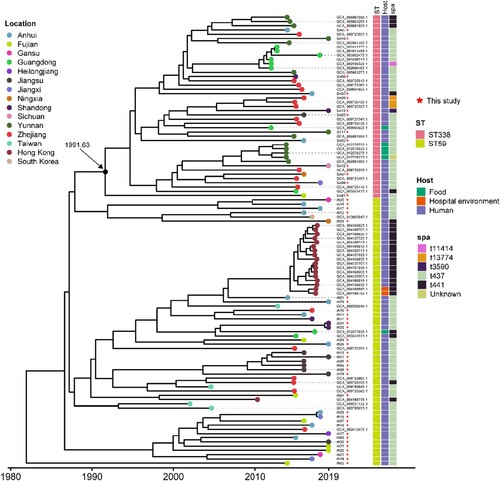
We constructed a phylogenetic tree of the 13 ST338 MRSA SCCmec Vb isolates and contemporaneous ST338 and ST59 MRSA SCCmec Vb isolates from China and adjacent geographic regions in order to estimate the time when the ST338 strain evolved from ST59. We identified 4150 SNPs in the alignment of 43 ST338 genome sequences (13 from this study and 30 from the NCBI database) and 63 ST59 sequences (33 from this study and 30 from the NCBI database); these were used to reconstruct the evolutionary history of the ST338 and ST59 lineages based on a Bayesian dating method in BactDating [Citation31]. The results showed that ST338 isolates probably evolved from ST59 isolates; the median time of emergence of the most recent common ancestor of all ST338 isolates was around 1991.63 (95% highest posterior density [HPD], 1984.39–2002.45) ().
Figure 2. Phylogeny of ST338 isolates. We aligned 43 ST338 genome sequences (13 from our study, 30 from that by NCBI database) and 63 ST59 sequence (33 from our database and 30 from NCBI). Isolate characteristics are shown on the right, including the location, host and Spa type.
We next calculated paired SNP distances between all ST338 SCCmec Vb MRSA isolates in this study and ST59 MRSA-SCCmec Vb isolates from China and other ST338 MRSA SCCmec Vb isolates in the NCBI database. The SNP difference between ST59 and ST338 ranged from 84 to 202 (median=152 and average=151) (Supplemental Figure S1). Notably, in addition to the SNP in the housekeeping gene gmk, there were 5 unique nonsynonymous SNPs that differentiated ST338 from the related ST59 clone; these were in genes related to methicillin resistance (fmtA) and metabolism of coenzymes (pdxt), lipids (uppS), and carbohydrates (odhA), and 1 SNP encoded a hypothetical protein (Table S2).
Virulence genes and pathogenicity islands in ST338
Virulence genes in the 13 ST338 isolates, rh10 (China mainland ST59-SCCmec Vb clone), M013 (Taiwan ST59-SCCmec Vb clone), MW2, and USA300 are shown in . In general, ST338 isolates shared the same virulence factors and genomic islands that were similar to those of M013 and rh10 but differed from those of MW2 and USA300. Unlike the latter 2 clones, all ST338 strains were negative for the clfA, clfB, eap, cna, sdrC, and sdrD genes encoding adhesion factors, suggesting a weaker adhesion capacity. ST338 isolates also had fewer exotoxin and enterotoxin genes.
Figure 3. Comparison of virulence factors between ST338, ST59 isolates, MW2 and USA300 strains. Red and white blocks represent the presence and absence of genes, respectively. The horizontal coloured bar represents (from left to right) genes associated with adhesion, serine protease, immune evasion, secretion system, enterotoxin, exfoliative toxin, leukocidin, toxic shock syndrome toxin, exotoxin, and pathogenicity islands.

All ST338 isolates harboured the vSaα, vSaβ, and vSaγ genomic islands. Unlike MW2 and USA300, none of the ST338 isolates had νSa3. vSaα encodes a set of putative staphylococcal exotoxin (set) genes, a lipoprotein gene (lpl), and a restriction/modification system (hsdM/hsdS). However, the set16, set17, set20, set21, and set23 genes present in the vSaα genomic island of MW2 were lacking in ST338 [(a)]. νSaβ encodes virulence factors such as serine proteases (splA–splF), enterotoxins, and the bicomponent toxin leukocidin LukDE; however, splA–splF and LukDE genes were absent in ST338. vSa3 contains sak (staphylokinase), sea (staphylococcal enterotoxin), sep (staphylococcal enterotoxin), and 2 new allelic forms of enterotoxin genes – ie, sel2 and sec4; none of the ST338 isolates harboured sak or sea (). The vSaγ genomic island encoding set, hla (α-toxin), extracellular fibrinogen-binding protein (efb), and psmβ (phenol-soluble modulin) is found in most S. aureus strains; α-toxin and psmβ are the major virulence factors of S. aureus.
Figure 4. Comparison of the vSaα and SaPI genomic islands between ST338 isolates (ST338-PVL+ and ST338-PVL-), M013 and rh10 (ST59), MW2 and USA300 strains. a, b. Comparison of vSaα (a) and SaPI (b) structure between ST338, rh10, M013, MW2 and USA300. Arrows and arrowheads represent open reading frames (ORFs) and their direction of transcription.
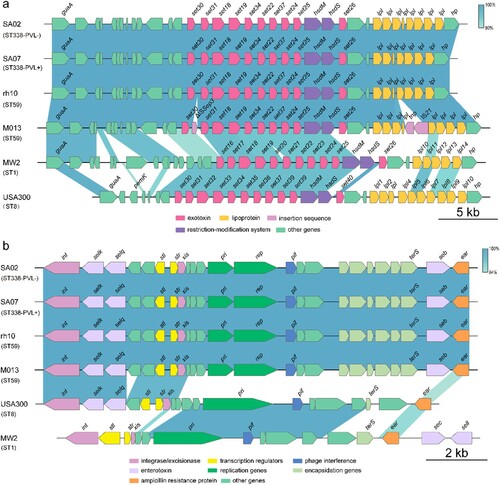
SaPIs are mobile genetic elements important for S. aureus evolution and virulence. All ST338 isolates harboured SaPI3, which contains seb, selk, and selq; in contrast, MW2 harbours sea, sec, and seh genes but lacks seb [(b)]. The ΦSa2 prophage – which carries lukF-PV and lukS-PV (PVL), the pore-forming cytotoxins associated with SSTIs and severe pneumonia caused by CA-MRSA [Citation37, Citation38] – was found in all but one ST338 isolate (SA02). PVL was not detected in SA02, suggesting that the high virulence of ST338 may be independent of PVL status.
The ΦSa3 prophage is not always occur in most of S. aureus, such as HA-MRSA strain COL (Gene bank number: CP000046.1). ΦSa3 carries chp (chemotaxis inhibitory protein), scn (complement inhibitor), sak (staphylokinase), and several enterotoxin genes (sea, seg2, sek2, and sep). In contrast to MW2, ST338 isolates did not contain sak or any enterotoxin genes, but harboured chp and scn (). Thus, the immune evasion cluster type C (ie, sak−) is present in νSaβ rather than in ΦSa3 in ST338 clones.
ST338 strains are less efficient in nasal colonization than MW2
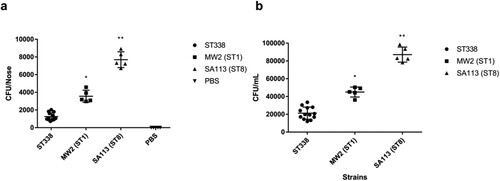
S. aureus mainly colonizes human nasal mucosa. The heatmap of virulence factors suggested that ST338 isolates may have weaker capacity for adhesion than MW2. We evaluated the nasal colonization capacity of the isolates in BALB/c mice with SA113 serving as a positive control, and confirmed that ST338 strains were less efficient in nasal colonization [P<0.01; (a)]; moreover, they had a lower capacity for epithelial cell adhesion than MW2 [P<0.05; (b)]. These results combined with the comparative analysis of virulence factors in ST338 and MW2 indicate that ST338 isolates are less efficient at host colonization than other CA-MRSA strains.
Figure 5. Nasal colonization and cell adhesion capacities of ST338 isolates. (a). Nasal colonization capacity of ST338 isolates compared to MW2 and SA113 strains in mice. Data for the thirteen ST338 isolates in five mice were averaged. (b). Adhesive capacity of ST338, MW2, and SA113 in A549 human alveolar epithelial cells. *P<0.05, **P<0.01.
Virulence of ST338
According to the case reports, the primary disease caused by ST338 in 9/13 cases was SSTI. To determine the virulence potential of ST338 in vivo, we used a mouse skin infection model. The MW2Δagr mutant strain served as a negative control and the low-virulence HA-MRSA ST239 strain was used for comparison. The size of skin abscesses was significantly larger in mice infected with ST338 isolates than in mice infected with MW2Δagr (p<0.05), but was comparable to that elicited by MW2 [(a)]. This indicates that the ST338-SCCmec Vb clone has strong potential to cause invasive skin infections.
Figure 6. The virulence potential of ST338 isolates in vivo. (a). Comparison of invasive capacity among ST338 isolates, MW2, HA-MRSA ST239 strain and the MW2Δagr mutant. Abscess area on day 3 after infection. **P<0.01 (q test). (b). Survival analysis of mice (n=10 per isolate) injected with 2×109 CFU or NaCl solution. Survival curves were compared with the log-rank (Mantel–Cox) test. (c). CFUs in kidneys. *P<0.05 (q test). (d). Haematoxylin and eosin staining of lung tissue at 48 h post infection. Inflammatory cell infiltration and tissue damage were greater in ST338 isolates than in HA-MRSA ST239 and the MW2Δagr mutant. A thickened alveolar septum, oedema and congestion, inflammatory cuffs of blood vessels, and leukocyte influx were observed.
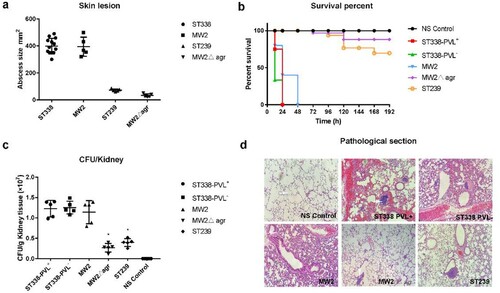
We also used a bacteraemia model to investigate the in vivo pathogenicity of ST338 isolates [(b) and Supplemental Figure S2a]. Mice infected with the PVL+ (SA01) and PVL− (SA02) ST338 strains all died with 24 h (n=10), while those infected with MW2 died within 48 h [(b)]. In all cases, infection with the MW2 and ST338 strains caused significantly greater mortality compared to the HA-MRSA strain ST239 and MW2Δagr mutant. The lungs of mice infected with the PVL+ and PVL− ST338 strains showed similar levels of tissue damage to those infected with ST239 and MW2Δagr [(d)]. Additionally, histopathologic examination of lung tissue samples revealed greater infiltration of inflammatory cells and tissue damage in the lungs of ST338-infected as compared to MW2-infected mice [(d)]. Mice infected with ST338 also showed greater inflammatory cell infiltration in the lungs than those infected with MW2, ST239, or MW2Δagr, with focal alveolar expansion and a thickened alveolar septum that was accompanied by higher CFU counts in the kidneys [(c)]. These findings demonstrate the high virulence of the ST338 isolates. It is worth noting that the virulence potential of the PVL− strain (SA02) was similar to that of other ST338 PVL+ strains, suggesting that the high virulence of ST338 strains mainly does not depend on their PVL status.
Core-genomic virulence genes are highly expressed in CA-MRSA ST338 isolates
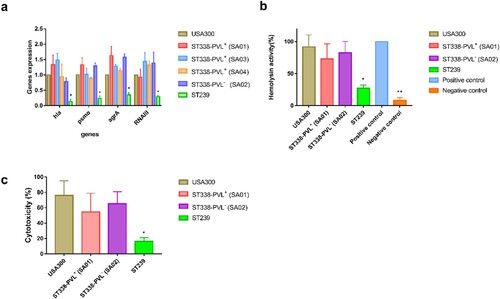
Given that the agr, hla, and psmα genes contribute to the virulence of CA-MRSA ST59 [Citation22], we investigated whether the high virulence of ST338 strains was due to upregulation of genome-encoded virulence factors. We examined the expression of genes encoding the toxins most frequently linked to CA-MRSA virulence including α-toxin (hla), PSMα peptides (psmα), agrA, and RNAIII in three PVL+ and one PVL− ST338 strain. All 4 genes were expressed at levels comparable to those in USA300 [P>0.05; (a)], although they were more highly expressed in ST338 isolates than in the ST239 strain. We also evaluated α-toxin production and cytotoxicity in ST338 and found that the isolates had significantly higher haemolytic capacity and cytotoxicity than HA-MRSA strains, but were comparable to USA300 in these aspects [(b,c)]. Thus, elevated expression of core virulence factors may be responsible for the high virulence of ST338 isolates.
Figure 7. Expression of virulence toxins and cytotoxicity of ST338 isolates. a. Expression of hla, psmα, agrA, and RNAIII in ST338 isolates compared to USA300 and ST239 strains. b. α-Toxin activity and production in ST338 isolates compared to USA300 and ST239 strains. c. Cytotoxicity of ST338 in human neutrophils compared to USA300 and ST239 strains. Values represent mean±SD of 3 independent experiments. *P<0.05, **P<0.01.
Discussion
The continuing increase of CA-MRSA infection rates worldwide is a major public health challenge [Citation39]. USA300 is the epidemic CA-MRSA clone in the US [Citation34], whereas ST59 is the most widespread clone in Asia [Citation22]. As a single-locus variant of ST59, ST338 has not yet been identified as a global pandemic clone, although it is increasingly detected in the Asia-Pacific region, especially in China [Citation15, Citation20, Citation40] where sporadic isolation of ST338-SCCmec Vb – mainly from children – has been reported [Citation41–43]. The complete genome of one ST338 strain has been published [Citation44].
In this study, we characterized 13 ST338-SCCmec Vb strains isolated from 13 severe bloodstream infection cases. The phylogenetic analysis indicated that the earliest ST338 isolate (SA07) may have acquired novel mutations to adapt to different environments. Furthermore, the phylogenetic reconstruction and time estimation suggested that ST338 evolved from ST59 in 1991. Notably, while SNP differences for isolates from the same clonal complex (CC) usually range from 0 to 3000 SNPs, differences between ST59 and ST338 ranged from 84 to 202 although both belong to CC59, indicating that the CA-MRSA ST338 SCCmec Vb isolates are closely related to the ST59 SCCmec Vb clone.
All ST338 isolates as well as ST59-SCCmec Vb strains harboured similar virulence factors and genomic islands; however, the former lacked the clfA, clfB, ebp, cna, sdrC, and sdrD genes found in MW2 and USA300. ClfA promotes bacterial colonization and adhesion [Citation45]; clfB, sdrC, and sdrD mediate adhesion to human nasal mucosa [Citation46–48]; and Cna is involved in tissue colonization [Citation49]. Experiments in mice and human alveolar epithelial cells showed that ST338 had a lower nasal colonization capacity than MW2, which is consistent with its lack of adhesion factor genes (clfA, clfB, ebp, sdrC, and sdrD).
The precise determinants of the high virulence potential of CA-MRSA strains are unclear [Citation12, Citation50]. It has been suggested that CA-MRSA but not HA-MRSA strains acquire the PVL-carrying mobile genetic element [Citation51], and that the expression of core genome-encoded toxin genes is upregulated in CA-MRSA strains [Citation52]. In this study, we analysed the virulence of ST338 isolates, of which twelve were PVL+ and one was PVL−, and found that they had comparable pathogenicity to MW2. We established mouse infection models to evaluate the invasive capacity of ST338 isolates; the results showed that ST338 strains were slightly more effective in causing SSTI than MW2, although the difference was nonsignificant; and the virulence potential of ST338 strains was comparable to that of MW2. Moreover, the virulence potential of the PVL− strain (SA02) was similar with that of PVL+ strains, implying that PVL status may be unrelated to the high virulence of ST338.
The significance of PVL status for CA-MRSA infection is still unclear. A highly virulent PVL− CA-MRSA strain (ST72) has been reported [Citation53], and the colonization ability of a PVL deletion mutant was found to be comparable to that of the parent strain in an in vivo infection model [Citation12]. We examined whether the high virulence potential of ST338 isolates depends on elevated expression of virulence factors using USA300 as a reference strain because of its high levels of related genes [Citation34, Citation35]. We found that the levels of hla, agrA, psmα, and RNAIII in ST338 isolates were comparable to those in the USA300 clone, demonstrating that the high expression of virulence factors in ST338 isolates may be responsible for their virulence, which was also supported by in vitro data.
In conclusion, this study provided the first detailed characterization of 13 ST338-SCCmec Vb CA-MRSA strains isolated from severe bloodstream infection cases. We demonstrated that the high virulence potential of these strains may be attributable to elevated expression of genome-encoded toxin genes. We also confirmed the emergence of a PVL− CA-MRSA ST338 strain that can cause severe SSTI comparable to PVL+ CA-MRSA strains. Our findings provide insight into the epidemiology of the novel and highly virulent ST338-SCCmec Vb clone. As the clinical and experimental data suggest that ST338-SCCmec Vb can cause serious and even fatal infections, its transmission should be monitored. However, because we did not investigate the factors contributing to the epidemiologic success of CA-MRSA isolates, it is difficult to predict whether ST338 CA-MRSA has epidemic potential like ST59.
Ethics statement
All procedures involving human participants and were performed in accordance with the ethical standards of First Affiliated Hospital of Zhejiang University, college of Medicine and with the 1964 Helsinki Declaration and its later amendments. All animal assays were approved by the Institutional Animal Care and Ethics Committee at The First Affiliated Hospital of Zhejiang University, College of Medicine.
Supplemental Material
Download (212.8 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- Otto M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev Dermatol. 2010;5:183–195. doi:https://doi.org/10.1586/edm.10.6.
- Tacconelli E, Tumbarello M, Cauda R. Staphylococcus aureus infections. N Engl J Med. 1998;339:2026–2027.
- Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol. 2014;17:32–37. DOI:https://doi.org/10.1016/j.mib.2013.11.004.
- Mediavilla JR, Chen L, Mathema B, et al. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr Opin Microbiol. 2012;15:588–595. DOI:https://doi.org/10.1016/j.mib.2012.08.003.
- Walraven CJ, Lingenfelter E, Rollo J, et al. Diagnostic and therapeutic evaluation of community-acquired methicillin-resistant Staphylococcus aureus (MRSA) skin and soft tissue infections in the emergency department. J Emerg Med. 2012;42:392–399. DOI:https://doi.org/10.1016/j.jemermed.2011.03.009.
- Otto M. Community-associated MRSA: a dangerous epidemic. Future Microbiol. 2007;2:457–459. DOI:https://doi.org/10.2217/17460913.2.5.457.
- Tracy LA, Furuno JP, Harris AD, et al. Staphylococcus aureus infections in US veterans, Maryland, USA, 1999-2008. Emerging Infect Dis. 2011;17:441–448. DOI:https://doi.org/10.3201/eid1703.100502.
- Naimi TS, LeDell KH, Como-Sabetti K, et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA. 2003;290:2976–2984. DOI:https://doi.org/10.1001/jama.290.22.2976.
- Herold BC, Immergluck LC, Maranan MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593–598. DOI:https://doi.org/10.1001/jama.279.8.593.
- Baba T, Takeuchi F, Kuroda M, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. DOI:https://doi.org/10.1016/s0140-6736(02)08713-5.
- Lee SM, Ender M, Adhikari R, et al. Fitness cost of staphylococcal cassette chromosome MEC in methicillin-resistant Staphylococcus aureus by way of continuous culture. Antimicrob Agents Chemother. 2007;51:1497–1499. DOI:https://doi.org/10.1128/AAC.01239-06.
- Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. DOI:https://doi.org/10.1146/annurev.micro.112408.134309.
- Li M, Cheung GY, Hu J, et al. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J Infect Dis. 2010;202:1866–1876. DOI:https://doi.org/10.1086/657419.
- Chambers HF. Community-associated MRSA--resistance and virulence converge. N Engl J Med. 2005;352:1485–1487. DOI:https://doi.org/10.1056/NEJMe058023.
- Chuang YY, Huang YC. Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Asia. Lancet Infect Dis. 2013;13:698–708. DOI:https://doi.org/10.1016/S1473-3099(13)70136-1.
- Wu D, Wang Q, Yang Y, et al. Epidemiology and molecular characteristics of community-associated methicillin-resistant and methicillin-susceptible Staphylococcus aureus from skin/soft tissue infections in a children's hospital in Beijing, China. Diagn Microbiol Infect Dis. 2010;67:1–8. DOI:https://doi.org/10.1016/j.diagmicrobio.2009.12.006.
- Li J, Wang L, Ip M, et al. Molecular and clinical characteristics of clonal complex 59 methicillin-resistant Staphylococcus aureus infections in mainland China. PloS one. 2013;8:e70602. DOI:https://doi.org/10.1371/journal.pone.0070602.
- Chen CJ, Unger C, Hoffmann W, et al. Characterization and comparison of 2 distinct epidemic community-associated methicillin-resistant Staphylococcus aureus clones of ST59 lineage. PloS one. 2013;8:e63210. DOI:https://doi.org/10.1371/journal.pone.0063210.
- Huang YC, Ho CF, Chen CJ, et al. Nasal carriage of methicillin-resistant Staphylococcus aureus in household contacts of children with community-acquired diseases in Taiwan. Pediatr Infect Dis J. 2007;26:1066–1068. DOI:https://doi.org/10.1097/INF.0b013e31813429e8.
- Liang B, Mai J, Liu Y, et al. Prevalence and Characterization of Staphylococcus aureus isolated from women and children in guangzhou, China. Front Microbiol. 2018;9:2790. DOI:https://doi.org/10.3389/fmicb.2018.02790.
- Huang YC, Ho CF, Chen CJ, et al. Comparative molecular analysis of community-associated and healthcare-associated methicillin-resistant Staphylococcus aureus isolates from children in northern Taiwan. Clin Microbiol Infect: Off Publ Eur Soc Clin Microbiol Infect Dis. 2008;14:1167–1172. DOI:https://doi.org/10.1111/j.1469-0691.2008.02115.x.
- Li M, Dai Y, Zhu Y, et al. Virulence determinants associated with the Asian community-associated methicillin-resistant Staphylococcus aureus lineage ST59. Sci Rep. 2016;6:27899. DOI:https://doi.org/10.1038/srep27899.
- Zheng B, Xu H, Yu X, et al. Low prevalence of MCR-1-producing Klebsiella pneumoniae in bloodstream infections in China. Clin Microbiol Infect: Off Publ Eur Soc Clin Microbiol Infect Dis. 2018;24:205–206. DOI:https://doi.org/10.1016/j.cmi.2017.08.004.
- Zheng B, Chen Y, Violetta L, et al. Bloodstream infections caused by entero-bacteriaceae in China. Lancet Infect Dis. 2019;19:810–811. DOI:https://doi.org/10.1016/S1473-3099(19)30352-4.
- Luo Q, Wan F, Yu X, et al. MDR salmonella enterica serovar typhimurium ST34 carrying mcr-1 isolated from cases of bloodstream and intestinal infection in children in China. J Antimicrob Chemother. 2020;75:92–95. DOI:https://doi.org/10.1093/jac/dkz415.
- Bioinformatics B. FastQC: a quality control tool for high throughput sequence data. Cambridge: Babraham Institute; 2011.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol J Comput Mol Cell Biol. 2012;19:455–477. DOI:https://doi.org/10.1089/cmb.2012.0021.
- Huang TW, Chen FJ, Miu WC, et al. Complete genome sequence of Staphylococcus aureus M013, a pvl-positive, ST59-SCCmec type V strain isolated in Taiwan. J Bacteriol. 2012;194:1256–1257. DOI:https://doi.org/10.1128/JB.06666-11.
- Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 2015;43:e15–e15.
- Didelot X, Croucher NJ, Bentley SD, et al. Bayesian inference of ancestral dates on bacterial phylogenetic trees. Nucleic Acids Res. 2018;46:e134–e134.
- Chen C. Searching for intellectual turning points: progressive knowledge domain visualization. Proc Natl Acad Sci USA. 2004;101(Suppl 1):5303–5310. DOI:https://doi.org/10.1073/pnas.0307513100.
- Wang Y, Liu Q, Liu Q, et al. Phylogenetic analysis and virulence determinant of the host-adapted Staphylococcus aureus lineage ST188 in China. Emerg Microbes Infect. 2018;7:45. DOI:https://doi.org/10.1038/s41426-018-0048-7.
- Kobayashi SD, Malachowa N, Whitney AR, et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 2011;204:937–941. DOI:https://doi.org/10.1093/infdis/jir441.
- Cheung GY, Wang R, Khan BA, et al. Role of the accessory gene regulator AGR in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79:1927–1935. DOI:https://doi.org/10.1128/IAI.00046-11.
- Jin Y, Li M, Shang Y, et al. Sub-Inhibitory concentrations of Mupirocin strongly inhibit alpha-toxin production in high-level Mupirocin-resistant MRSA by down-regulating agr, saeRS, and sarA. Front Microbiol. 2018;9:993. DOI:https://doi.org/10.3389/fmicb.2018.00993.
- Yoong P, Torres VJ. The effects of Staphylococcus aureus leukotoxins on the host: cell lysis and beyond. Curr Opin Microbiol. 2013;16:63–69. DOI:https://doi.org/10.1016/j.mib.2013.01.012.
- Gillet Y, Issartel B, Vanhems P, et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359:753–759. DOI:https://doi.org/10.1016/S0140-6736(02)07877-7.
- Dantes R, Mu Y, Belflower R, et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013;173:1970–1978. DOI:https://doi.org/10.1001/jamainternmed.2013.10423.
- Peng H, Liu D, Ma Y, et al. Comparison of community- and healthcare-associated methicillin-resistant Staphylococcus aureus isolates at a Chinese tertiary hospital, 2012-2017. Sci Rep. 2018;8:17916. DOI:https://doi.org/10.1038/s41598-018-36206-5.
- Geng W, Yang Y, Wang C, et al. Skin and soft tissue infections caused by community-associated methicillin-resistant Staphylococcus aureus among children in China. Acta Paediatr. 2010;99:575–580. DOI:https://doi.org/10.1111/j.1651-2227.2009.01645.x.
- Geng W, Yang Y, Wu D, et al. Molecular characteristics of community-acquired, methicillin-resistant Staphylococcus aureus isolated from Chinese children. FEMS Immunol Med Microbiol. 2010;58:356–362. DOI:https://doi.org/10.1111/j.1574-695X.2010.00648.x.
- Geng W, Yang Y, Wu D, et al. Community-acquired, methicillin-resistant Staphylococcus aureus isolated from children with community-onset pneumonia in China. Pediatr Pulmonol. 2010;45:387–394. DOI:https://doi.org/10.1002/ppul.21202.
- Chen Y, Hong J, Chen Y, et al. Characterization of a community-acquired methicillin-resistant sequence type 338 Staphylococcus aureus strain containing a staphylococcal cassette chromosome mec type VT. Int J Infect Dis: IJID: Off Publ Int Soc Infect Dis. 2020;90:181–187. DOI:https://doi.org/10.1016/j.ijid.2019.10.034.
- Herman-Bausier P, Labate C, Towell AM, et al. Staphylococcus aureus clumping factor A is a force-sensitive molecular switch that activates bacterial adhesion. Proc Natl Acad Sci USA. 2018;115:5564–5569. DOI:https://doi.org/10.1073/pnas.1718104115.
- O'Brien LM, Walsh EJ, Massey RC, et al. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell Microbiol. 2002;4:759–770. DOI:https://doi.org/10.1046/j.1462-5822.2002.00231.x.
- Feuillie C, Formosa-Dague C, Hays LM, et al. Molecular interactions and inhibition of the staphylococcal biofilm-forming protein SdrC. Proc Natl Acad Sci USA. 2017;114:3738–3743. DOI:https://doi.org/10.1073/pnas.1616805114.
- Ajayi C, Aberg E, Askarian F, et al. Genetic variability in the sdrD gene in Staphylococcus aureus from healthy nasal carriers. BMC Microbiol. 2018;18:34. DOI:https://doi.org/10.1186/s12866-018-1179-7.
- Rhem MN, Lech EM, Patti JM, et al. The collagen-binding adhesin is a virulence factor in Staphylococcus aureus keratitis. Infect Immun. 2000;68:3776–3779. DOI:https://doi.org/10.1128/iai.68.6.3776-3779.2000.
- Otto M. A MRSA-terious enemy among us: end of the PVL controversy? Nat Med. 2011;17:169–170. DOI:https://doi.org/10.1038/nm0211-169.
- Tristan A, Bes M, Meugnier H, et al. Global distribution of Panton-Valentine leukocidin--positive methicillin-resistant Staphylococcus aureus, 2006. Emerging Infect Dis. 2007;13:594–600. DOI:https://doi.org/10.3201/eid1304.061316.
- Wang R, Braughton KR, Kretschmer D, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. DOI:https://doi.org/10.1038/nm1656.
- Chen Y, Yeh AJ, Cheung GY, et al. Basis of virulence in a Panton-Valentine leukocidin-negative community-associated methicillin-resistant Staphylococcus aureus strain. J Infect Dis. 2015;211:472–480. DOI:https://doi.org/10.1093/infdis/jiu462.