
ABSTRACT
Highly pathogenic avian influenza viruses (HPAIVs) frequently receive global attention as threats to public health. The NS1 protein is a key virulence factor known to impair host antiviral responses. The study herein revealed HPAIV H5N2 NS gene encoded additional protein; a truncated NS1 variant, designated NS3, produced by alternative splicing of the NS transcript. To examine the function of NS3 during infection, we generated recombinant viruses expressing either full-length NS1 (RG-AIV-T375G) or NS3 (RG-AIV-NS3). Interestingly, RG-AIV-NS3 virus produced higher titres than RG-AIV-T375G in multiple mammalian cell lines. However, RG-AIV-T375G exhibited a replication advantage over RG-AIV-NS3 in chicken DF-1 cells, indicating that host cell identity dictates the effect of NS3 on viral replication. In mice and mammalian cells, RG-AIV-NS3 infection elicited higher level of cytokines, including IFN-β, MX and TNF-α, potentially due to its higher replication activity. Based on mini-genome assay, NS3 had pronounced effects on viral replication machinery. Surprisingly, NS3 retained an interaction with PKR and suppressed PKR activation despite its lack of amino-acid residues 126-167. The poor replication ability of RG-AIV-T375G was partially restored in cells deficient in PKR suggesting that full-length NS1 may be insufficient to suppress PKR function. Notably, virulence of the full-length NS1-expressing RG-AIV-T375G virus was highly attenuated in mice when compared to RG-AIV-NS3. In summary, our study reveals the existence and function of a previously unidentified H5N2 viral protein, NS3. We found that NS3 is functionally distinct from NS1 protein, as it enhances viral replication and pathogenicity in mammalian systems, potentially via suppression of PKR activity.
Introduction
Many avian influenza viruses (AIVs) can infect a variety of species and may pose severe threats to public health. For example, the highly pathogenic AIV (HPAIV) H5N1 was first reported as an infection in domesticated geese in southern China in 1996, and since its identification, H5N1 has continually caused outbreaks in animals and humans across Asia, the Middle East, North America and Africa [Citation1,Citation2]. Unlike the HPAIVs, which frequently cause devastating effects on the poultry industry, low pathogenic AIVs (LPAIVs) usually causes mild disease or asymptomatic infections in poultry. In Taiwan, the LPAIV H6N1 was first isolated in 1972 [Citation3], and it has remained a dominant endemic strain since that time [Citation4]. Nevertheless, several AIVs of the H5 subtype have emerged in Taiwan within the last two decades. The first outbreak of LPAIV H5N2 in Taiwan was reported in 2003, followed by a second wave in 2008 and subsequent occasional detection in chickens [Citation4,Citation5]. Sequence analysis implied that the virus (also called Mexican-like H5N2) is the result of reassortment events. As such, the viral genome harbors HA and NA gene segments that originated from an H5N2 strain first isolated in Mexico and another six internal genes that were inherited from local enzootic H6N1 viruses [Citation5]. As a result of its co-circulation with H6N1, the Mexican-like H5N2 virus evolved into an HPAIV in 2012; this HPAIV retains the internal gene complex from the H6N1 virus lineage but has several modifications from multiple reassortment events [Citation6]. Moreover, in early 2015, three novel H5-clade 2.3.4.4 HPAIVs emerged. These emergent H5 viruses included the Eurasian lineage of H5N2, H5N8 and H5N3, which all caused high mortality in geese [Citation7]. Soon after this episode, another novel HPAIV (H5N6) was detected in a dead duck, and this strain caused sporadic outbreaks throughout 2017 [Citation8]. Such continuous evolution and emergence of new subtypes along with sustained transmission of AIVs in the domestic poultry industry creates a need to continuously monitor the status of H5 epidemics and to better understand the pathogenic mechanisms of these viruses.
The genome of influenza A virus is composed of eight RNA segments encoding at least ten major viral proteins. In order to generate the full set of proteins, two genomic segments (numbers 7 and 8) give rise to distinct transcripts by alternative splicing [Citation9]. The splicing of segment 8 creates mRNAs that encode nonstructural (NS) protein 1 (NS1) and NS2. In addition to the generation of multiple mRNAs by alternative splicing, the canonical protein and additional viral proteins may arise from a single mRNA by effects on translation machinery. For instance, leaky ribosomal scanning will lead to the translation of PB1-F2 from an alternative reading frame (+1 open reading frame) of the PB1 gene, using a downstream AUG [Citation10]. Moreover, two N-terminus-truncated NS1 proteins were found to be translated from AUGs located at positions 235 and 241 of the NS1 open reading frame [Citation11]. Importantly, these truncated viral proteins can affect the replication of influenza A viruses in a strain-specific manner.
The NS1 protein is translated from the NS transcript without splicing. The protein forms a dimer and contains two functional domains, including a double-stranded RNA (dsRNA)-binding domain (RBD) and an effector domain (ED) respectively located at the N- and C-terminus of the protein [Citation12]. In terms of function, NS1 is highly pleiotropic and widely regarded as a key determinant of virulence. The protein is also essential to viral fitness during the course of interspecies adaptation, principally due to its roles in counteracting innate immune response and in modulating expression machinery activities [Citation13]. It is well documented that host cell detection of influenza virus RNA triggers expression of type I interferon (IFN) and proinflammatory cytokines by cytosolic RIG-I and endosomal Toll-like receptors (e.g. TLR3 and TLR7) [Citation14]. However, NS1 restricts IFN signalling via several mechanisms that involve the interplay of NS1 with multiple cellular partners. Both the RBD and ED of NS1 contribute to the suppression of IFN response. The RBD associates with dsRNA, a potent inducer of type I IFN, and directly interacts with the second CARD (caspase activation and recruitment domain) of retinoic acid-inducible gene I (RIG-I) [Citation15,Citation16]. These actions ultimately lead to the suppression of IFN expression over the course of viral infection [Citation17]. Moreover, NS1 impairs IFN-mediated antiviral responses downregulating IFN signalling pathways and modulating cellular mRNA maturation machinery, an effect mediated by intermolecular interactions between the ED region of NS1 and its cellular targets (e.g. PKR, TRIM25 and CPSF30) [Citation17–20].
High variation has been noted within the NS sequences of H5N2 strains that have emerged in Taiwan over the last decade. Sporadic mutations spanning the NS1 coding region have been identified, causing sequence similarities between major strains to be as low as 90.6% [Citation21]. Importantly, substitution of heterologous NS genes can markedly affect the infection efficiency of reassortant viruses and the cytokine expression profiles of infected cells, supporting a crucial role for NS1 in both viral compatibility and host immune responses. Of note, the HPAIV H5N2 strain (A/goose/Taiwan/01031/2015) NS1031 gene was found to encode two proteins, including NS1 and additional protein with smaller size [Citation21]. Based on its molecular weight, the smaller product is unlikely to be an N-terminus-truncated NS1 initiated at an internal AUG codon (position 235 or 241) that was identified in a previous report [Citation11]. Since the reassortant virus bearing the NS1031 gene is greatly attenuated in mammalian cells [Citation21], we sought to investigate the identity and characteristics of the shorter NS1031-encoded protein in the context of influenza infection. In this study, we found that the NS1031 virus encodes a variant NS1 protein, denoted as NS3 based on previous study on H3N2 virus [Citation28]. NS3 protein is translated from an alternatively spliced transcript. We then went on to compare the roles of NS3 with those of full-length NS1. According to our results, the NS3 protein appears be functionally non-redundant with the NS1 protein. Despite its internal deletion, NS3 enhances viral replication and pathogenicity of influenza virus in both cell and animal models. Moreover, it effectively suppresses PKR activity and confers a growth advantage to influenza virus in mammalian cells.
Materials and methods
Virus and cells
H1N1 virus strain A/Puerto Rico/8/1934 (PR8) and reassortant viruses that express, wild type NS gene from the 1031 strain (A/goose/Taiwan/01031/2015), or only one of the two NS1 variants were propagated in 10-day-old embryonated specific-pathogen-free (SPF) chicken eggs for 48 h at 37°C. Cells used in this study were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco BRL, Life Technologies Corporation Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS, Hyclone, Logan, UT, USA) and antibiotics.
Construction of plasmids for reverse genetics
Two reassortant viruses expressing one of the two NS1 variants originated from NS segment (accession number KU646889.1) of A/goose/Taiwan/01031/2015 avian influenza virus (abbreviated as strain 1031 herein), were generated by reverse genetics. Initially, the NS segments bearing sequences expressing either the full length NS1 (T375G) or NS3 were produced by PCR. DNA expressing only the full length NS1 was obtained by introducing a T-to-G point mutation at nucleotide 375 via site-directed mutagenesis that resulted in the replacement of the novel splicing donor dinucleotide (GU) with GG. While, the DNA bearing NS3 sequences was reconstituted from NS1031 gene by overlapping extension PCR. Primers were available in Supplementary Table 1. To construct reverse genetic vectors, the resulting PCR product was then subcloned into reverse genetic vector pDZ kindly provided by Professor Peter Palese (Icahn School of Medicine at Mount Sinai, USA) [Citation22], following the strategies described in our previous report [Citation21].
Rescue of reassortant AIVs
HEK293T cells were transfected with the mixture of pDZ plasmids containing eight gene segments (seven gene segments derived from PR8 strain and the NS encoding one of the NS01031 isoforms) by Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA). After 24-hr incubation, cells were harvested and inoculated into 10-day-old SPF eggs for viral amplification. The progeny viruses in allantoic fluid were further purified by plaque assay in MDCK cells. Three single plaques of each reassortant virus were picked and further propagated in SPF eggs. Sequences of NS segments were initially confirmed by automated sequencing (Mission Biotech. Co. Ltd., Taiwan), and whole genome sequences of the purified viruses were further validated by next generation sequencing (Genomics Biotech. Co. Ltd., Taiwan).
Generation of constructs expressing various NS1 isoforms
Two sets of constructs were generated for expression of NS1 proteins with fusion of either an FLAG-tag or eGFP protein at its C-terminus. PCR was used to amplify the coding region of the four NS1 variants. Primers were available in Supplementary Table 1. The resulting PCR fragments were subsequently treated with two restriction enzyme sets, EcoR I/Kpn I or Bgl II/Not I, for cloning into vector pCMV14 (Sigma-Aldrich) or GFP-pcDNA3.1 [Citation23], which expresses an FLAG-tag and eGFP fusion protein, respectively.
Plaque assay
MDCK cells seeded in 12-well plates were washed with PBS once and infected with 400 μL/well of viruses that were serially diluted in the infectious medium (DMEM with TPCK-treated trypsin (1.0 µg/mL)). At three hours post infection (hpi), after removal of infectious medium, cells were washed with PBS and then overlaid with infectious medium containing of 0.6% agarose. At 48 hpi, cells were fixed by 10% formaldehyde and plaques were visualized by crystal violet stain. The virus titre was estimated and expressed as the mean plaque-forming units (PFU) per mL.
Growth kinetics of reassortant AIVs
Cells were infected with the reassortant viruses, at a multiplicity of infection (MOI) of 0.01 for 12, 24, 36, and 48 hpi. The titres of viral progenies in the culture medium were measured by standard plaque assay.
Transfection
Transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Briefly, cells were seeded in a 12-well plate one night prior to transfection to reach approximately 80% confluency. In total, 2 μg of plasmids were mixed with 4 μL of liposome that was diluted in 100 μL of DMEM (without FBS or antibiotics) at room temperature. After 20-min incubation, the mixture of DNA and liposome was added dropwise onto cell monolayers and incubated for 1 d for further analysis.
Western blot analysis
Whole cell lysates were prepared and separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) followed by electro-transfer onto nitrocellulose (NC) paper (Bio-Rad). The filter was blocked in 5% skim milk for one hr, and then incubated with the primary antibodies at 4°C for overnight. Subsequently, the filter was rinsed with PBS containing 0.05% Tween 20 (PBS-T) for five times, followed by incubation with the corresponding secondary antibody conjugated with horseradish peroxidase (HRP) for 1 hr. After washing with PBS-T, protein signals were detected by enhanced chemiluminescence (ECL) and acquired by ImageQuant LAS 4000 (GE Healthcare, Uppsala, Sweden). The dilutions of each antibody were as follows: anti-β-actin (1:1,000; Signalway Antibody), anti-FLAG (1:2,500; Signalway Antibody), anti-β-actin (1:5,000; Jackson), anti-NS1 (1:2,000, GeneTex), anti-NP (1:1,000; GeneTex), anti-PKR (1:2,000, Abcam), anti-PKR-p (T446; 1:2,000; Abcam), anti-HA (1:2,500; Yao-Hong Biotechnology, Taiwan).
Ratio of NS3/NS1 mRNA and protein
Total cellular RNA was extracted by the Trizol® method (Invitrogen, Carlsbad, CA, USA). The RT-qPCR method was used to measure and estimate the ratio of NS3/NS1 mRNA. For relative quantification, expression levels of NS3/NS1 mRNA from three independent experiment were normalized by 2−△Cq with full length NS segment. Primers used to analysis splicing ratio were listed in Supplementary Table 2.
Moreover, ratio of NS3/NS1 protein was estimated by quantification of the signal from western blot analysis by Image-J software.
Immunofluorescence assay (IFA)
Transiently transfected cells were fixed with 1.875% formaldehyde for 10 min, followed by permeabilization with 0.5% NP-40, and then incubation with anti-NS1 antibody (sc-130568, Santa Cruz Biotechnology) at room temperature for 1 hr. After rinse with PBS containing 1% FBS, cells were further incubated with corresponding secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA) for an additional 1 hr at room temperature. Subsequently, cells were stained with DAPI (4′, 6-diamidino-2-phenylindole) at a final concentration of 1 µg/mL. Images were acquired by confocal microscopy (FV1000, Olympus, Tokyo, Japan) with Olympus FV10-ASW 1.3 viewer software.
Mini-genome reporter assay
Human 293 T cells seeded in 48-well plates were transfected with constructs expressing viral RNA-dependent RNA polymerase (RdRp) complex derived from H1N1 (PR8 strain) or H5N2 (NS1031 strain), including NP (20 ng), PB1 (20 ng), PB2 (20 ng), PA (20 ng), and a firefly luciferase expression plasmid (10 ng) as an internal control for transfection efficiency, as well as a reporter plasmid, pPol I-Flu-Rluc (100 ng) [Citation24]. Plasmid pPol I-Flu-Rluc carries a mini-genome that contains Renilla luciferase and the initial RNA transcription was driven by human RNA polymerase I promoter, while the subsequent expression of Renilla luciferase was controlled by influenza polymerase complex. At 24 hr post-transfection, the luminescence in transfected cells was analysed using a Dual-Glo luciferase assay system following manufacturer instructions (Promega). Luciferase activity was then acquired by a FLUOstar OPTIMA microplate reader (BMG Labtech GmbH, Offenburg, Germany). Relative light units (RLU) were estimated as the ratio of Renilla to firefly luciferase luminescence. Relative RdRp activity was calculated as a percentage of the maximum RLU in each experiment.
Immunoprecipitation (IP)
Total proteins were harvested from transfected cells expressing each of the FLAG-tagged NS1 isoforms in lysis buffer containing proteinase inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). After clarification by centrifugation, one-tenth of the whole-cell lysates was kept as the input control. The remaining cell lysates was then reacted with anti-FLAG M2 affinity gels (Sigma-Aldrich, St. Louis, MO, USA) at 4°C for overnight. Subsequently, the gel was washed thoroughly with wash buffer (50 mM Tris, pH 7.4 and 150 mM NaCl) and target proteins were then eluted in SDS sample dye. The interaction profile was revealed by western blot analysis.
Quantification of cytokine transcripts with quantitative real-time RT–PCR (qRT–PCR)
The effect of recombinant AIVs on cytokine expression was evaluated in various cell types and also the mouse model. First, cells were infected with individual viruses at an MOI of 0.1 in three independent repeats. At 6 and 12 hpi, total RNA was isolated from cells by Trizol® (Invitrogen, Carlsbad, CA, USA) and processed by TURBO DNA-Free kit (Invitrogen). Moreover, 5 six-week-old BALB/c mice in each group were intratracheally inoculated with 1 × 105 PFU (in 50 μL) of the reassortant viruses. At 12 and 24 hpi, lungs of the infected mice were collected and 0.1∼0.2 g of lung was then homogenized by TissueLyser II (QIAGEN, Netherlands). Total RNA of tissue was extracted by Maxwell RSC simplyRNA Tissue kit (Promega). The experimental protocol and sample collection of animals had been approved by the Institutional Animal Care and Committee of National Chung Hsing University (IACUC number: 107-148).
Cytokine expression was monitored by one-step RT-qPCR using gene specific primers (summarized in Supplementary Table 2). RNA was reverse-transcribed with GoTaq 1-Step RT-qPCR System kit (Promega) followed by cytokine quantification (CFX ConnectTM Real-Time PCR Detection System, Bio-Rad). Expression data from three independent experiment were relativized by 2−△△Cq with endogenous β-actin standard, and all values were estimated as fold above mock group. Moreover, viral M RNA segment was quantified following the method described in one previous report [Citation25].
Quantification of viral M RNA yield and cytokine level in infected mice
Serum samples were collected from infected mice at 12 and 24 hpi. Total RNA was extracted by Maxwell RSC simplyRNA Tissue kit (Promega). Subsequently, RNA of viral M segment was quantified following the method described in one previous report [Citation25]. In addition, IFN-β concentrations in serum of each IAV-infected mouse was determined by commercial ELISA (VeriKineTM Mouse Interferon Beta ELISA Kit, PBL Assay Science, Piscataway, NJ, USA) following the manufacturer's instruction.
Pathogenicity study in the mouse model
Five-week-old female BALB/c mice (n = 7) were intranasally inoculated with 105 PFU of the reassortant viruses. Body weights and survival were monitored daily for two weeks. The experimental protocol was approved by the Institutional Animal Care and Committee of National Chung Hsing University (IACUC number: 107-148).
Statistical analysis
The growth kinetics between groups was comparatively analysed by Two-way ANOVA, and the data were displayed as mean ± standard deviation. Differences of cytokine expression level among groups were evaluated by SAS (SAS Institute, Cary, North Carolina, USA) using the unpaired student t-test. The ratio of NS3/NS1 mRNA and protein was analysed by One-way ANOVA. The statistical analysis in pathogenicity study was performed using method of Kaplan Meier with a Log-rank test. All data were analysed using GraphPad Prism 5 software (GraphPad Software, San Diego, California, USA). P-values less than 0.05 were considered statistically significant.
Results
Identification of NS3 protein encoded from hpaiv H5N2
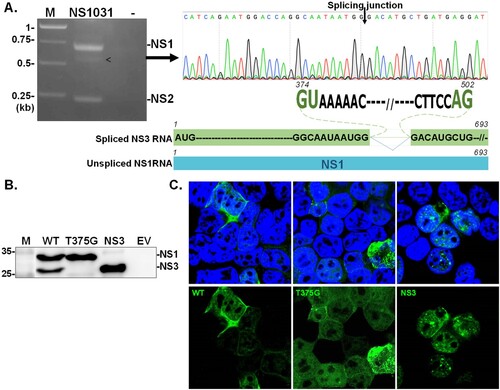
First, we sought to identify the NS1 variant in the H5N2 1031 strain. As shown in (A), three NS fragments could be amplified by RT–PCR from the RNA of 1031 strain-infected cells. Sequence analyses on each fragment revealed a previously unidentified NS transcript bearing an internal deletion spanning from nucleotide (nt) 374–502 in the NS1 coding region ((B), indicated by arrowhead). Of note, the conserved splicing dinucleotide (GT-AG) of the intron was present at the termini of the deleted region, suggesting that the novel NS transcript (designated NS3) resulted from an alternative splicing event on NS1 RNA. This assumption was confirmed by eliminating the splicing donor dinucleotide; T375 was substituted with G (a silent mutation). As expected, the mutant NS1031(T375G) expressed only full-length NS1; meanwhile, the construct bearing NS3 sequences encoded a protein with a size representing the smaller NS1 variant ((C)). Moreover, the T375G-encoded full-length NS1 and NS3 proteins were expressed at high levels and distributed throughout the majority of positive-staining cells (59% full-length and 77% NS3). Of note, in cells with lower expression levels, full-length NS1 was mainly localized in the cytoplasm (38% of positive cells), while NS3 was generally localized to the nucleus (21% of positive cells) ((D)).
Figure 1. Identification of NS3. NS transcripts were amplified by RT–PCR from cells infected with RG-AIV-WT virus (A). The identity of an unexpected transcript (indicated by arrowhead) was revealed by automated sequencing. Alignment analysis indicated that the additional transcript, denoted as NS3, was a spliced version of NS1. NS3 contained sequences corresponding to NS1 with a deletion between the splicing dinucleotides 374GU-AG502. The expression of NS1 isoforms from NS1031 was validated (B–C). The expression profiles and cellular distributions of proteins expressed from wild-type NS1031 (WT), a construct with a substitution of splicing donor site (T375G), and the NS3 construct (deletion of NS1 residues 375-502) in transiently transfected cells were examined by western blot analysis (B) and fluorescence microscopy (C).
NS3 and t375g direct the growth preference of influenza virus in different host cells
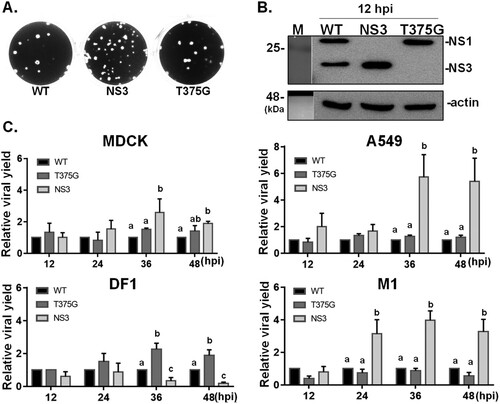
To assess the function of the NS3, two recombinant influenza viruses were generated by a reverse genetic technique. RG-AIV-T375G was made to express full-length NS1 (T375G), while RG-AIV-NS3 expressed NS3. We first compared the growth kinetics and viral protein expression of these reassortant viruses with another virus bearing the wild-type NS1031 gene (RG-AIV-WT; expresses both full-length NS1 and NS3). The three viruses all yielded plaques with similar sizes ((A)) and expressed the expected NS proteins ((B)). Interestingly, the internal deletion of 43 amino acids in the NS protein (encoded by the RG-AIV-NS3 virus) had significantly higher yields in mammalian cells (MDCK, A549 and M1 cells) compared to both RG-AIV-WT and RG-AIV-T375G ((C)). In contrast, the yield of RG-AIV-T375G was significantly higher than the other two viruses in chicken DF-1 cells, suggesting that the NS1 variants differentially influenced the infection efficiencies in different host cell contexts ((C)).
Figure 2. Growth kinetics of reassortant viruses expressing various isoforms of NS1031. Recombinant AIVs bearing the NS segments derived from NS1031. Constructs were made for expressing wild-type NS1 (WT), only full-length NS1 (T375G), or NS3 (NS1 with internal deletion). Infection with these three reassortant viruses produced plaques of similar sizes (A) and the expected NS1 profile (B) in MDCK cells. Growth kinetics of the three reassortant viruses in four cell lines: human A549, canine MDCK, mouse M1 cells, and chicken DF1 cells. The titres of viral progenies were measured from the four cell lines (infected with 0.01 MOI of the indicated virus) after 12, 24, 36 and 48 h. The viral yields of each variant were plotted relative to wild-type virus (C).
We also monitored the levels of viral proteins, including NP and NS1 in the infected cells. As early as 6 hpi, expression of the NS1 variants was readily observed in DF1 cells ((A)). However, in mammalian cells infected with RG-AIV-NS3 virus, the NP levels were significantly higher than those in corresponding cell lines infected with either of the other two viruses at 12 hpi ((B), upper panel). Among the three mammalian cell types, M1 cells had significantly higher NS3 levels after infection with RG-AIV-NS3 compared to RG-AIV-WT ((B) lower panel). Of note, NS3 expression was apparently lower than NS1 expression in RG-AIV-WT-infected M1 cells ((A)); hence we calculated the ratios of these two variants in other contexts. Interestingly, the ratio of NS3/NS1 protein levels was significantly lower in M1 cells than in the other two mammalian cell types ((C), upper panel); yet, the ratios of NS3/NS1 mRNA level showed no differences among the three cell types ((C), lower panel).
Figure 3. Expression levels of NS1 variants in infected mammalian and avian cells. Levels of accumulated viral proteins, including NP, NS1 and NS3, in mammalian (A549, and M1) and avian DF1 cells were determined by immunoblot analysis after infection with the indicated viruses for 6 and 12 h (A). The level of viral protein expressed by RG-AIV-WT was arbitrary set as 1, and the relative expression levels of viral proteins were plotted for different viruses at 12 hpi (B). The ratio of NS3/NS1 protein (C) and RNA (D) were determined from different cell types infected by indicated viruses. *p < 0.05.
NS3 enhances viral genome replication
The aforementioned results revealed a trend in which NS1 variant expression patterns ((B)) correspond with viral yield in all three analyzed mammalian cell lines ((C)). In addition to counteracting innate immune response, NS1 is known to influence viral RNA dependent RNA polymerase (RdRp) complex activity [Citation21]. Thus, we further investigated whether the efficient replication of RG-AIV-NS3 virus might involve NS3 effects on viral RdRp activity. The genetic backbone of the RG-AIV-NS3 virus was based on H1N1 (strain PR8), so we monitored the activity of PR8-derived RdRp by means of a mini-genome assay. As shown in (A), the PR8 NS1 protein strongly enhanced RdRp activity. Similarly, in the presence of NS3, the reporter activity driven by the RdRp complex of PR8 virus was significantly increased, while neither wild-type NS1 nor full-length NS1 (T375G) influenced viral genome reporter activity. Nevertheless, none of the NS1 variants could enhance RdRp complex activity when derived from the H5N2 virus (A/goose/Taiwan/01031/2015), the parental virus of the NS1031 gene ((B)).
Figure 4. Impacts of NS1 variants on viral genome activity. (A) The effect of NS1 isoforms on viral RNA dependent RNA polymerase (RdRp) activity was evaluated by mini-genome reporter assay. HEK293T cells were co-transfected with one of the NS1 plasmids (PR8, WT NS1031, NS3 or T375G) along with constructs expressing the viral RdRp complex (PA, PB1, PB2 and NP proteins); the RdRp complex expression construct was based on either PR8 (A), or NS1031 virus (B). In addition to these two plasmids, a reporter plasmid pPol I-Flu-Rluc and internal control plasmid expressing firefly luciferase were also transfected. At 24 h post-transfection, luciferase activities (top panel) and NS1 expression (bottom panel) of the whole cell lysate were respectively measured with the Dual-Glo luciferase assay and western blot analysis. The Renilla luciferase expression level was initially normalized with firefly luciferase activity in the same group. Then, the relative expression level was obtained by comparison with the EV control, which was arbitrarily set as 1. The experiment was conducted in triplicate. *p < 0.05.
NS3 interacts with and suppresses activation of PKR
Since the molecular function of NS3 was still unknown, we investigated whether the deletion of residues 126–168 affects the known effect of NS1 on suppressing PKR activation. Cells were made to transiently express both proteins (i.e. WT), or only full-length NS1 (T375G) or NS3, and PKR activity was monitored according to its phosphorylation level. Surprisingly, NS3 significantly suppressed the activation of PKR, while T375G or WT only mildly influenced PKR activation status ((A,B)). It is well documented that NS1 suppresses PKR activity via a direct intermolecular interaction, so we performed immunoprecipitation (IP) to test the interaction level. Despite the internal deletion, NS3 alone pulled down PKR to a greater extent than WT or T375G ((C,D)).
Figure 5. Effects of NS1 variants on protein kinase R (PKR) activation and interaction. NS1 modulates PKR activation. HEK293T cells were transfected with empty vector (EV), or plasmid expressing an NS1 protein variant (WT, T375G or NS3), followed by stimulation of PKR activation by the transfection of 150 ng/ml poly I:C for 4 hr. The phosphorylation level of PKR was monitored by Western blot analysis (A), and PKR activation level relative to EV was estimated and plotted (B). NS1 variants interact with PKR. HEK293T cells were transfected with plasmids expressing hemagglutinin (HA)-tagged PKR together with either EV or one of the constructs expressing an NS1 isoform. After transfection, FLAG-IP and immunoblotting were performed (C). The level of PKR-NS1 interaction relative to WT NS1031 was estimated and plotted (D). Replication efficiency of reassortant RG-AIVs in A549 cells deficient in PKR function was determined (E). Human A549 cells without (WT) or with knockout (KO) of PKR function were infected with reassortant RG-AIVs expressing WT NS1, NS3, or T375G for 12, 36 and 48 hpi. The titres of virus progenies were determined by the standard plaque assay. The titre of each virus in PKR-WT cells was set as 1, and the relative viral yields in infected PKR-KO cells were then calculated. All experiments were conducted in triplicate. *p < 0.05, **p < 0.01.
Intriguingly, full-length protein T375G appeared to only weakly interact with PKR and had a minor if any effect on PKR activation. To further explore the relevance of this finding, the three viruses were used to infect A549 cells with knockout of PKR expression [Citation26]. The viral titre of RG-AIV-T375G was significantly increased in infected PKR-KO cells compared to wild-type A549 cells at 36 hpi; however, the yields of RG-AIV-WT or RG-AIV-NS3 were similar in both cell types ((E)). Of note, the same was also true at the later time point of 48 hpi.
NS3 stimulates cytokine response and promotes pathogenicity in mammalian models
NS1 is regarded as a major determinant of influenza virulence, so we wanted to compare the effects of full-length NS1 and NS3 on the pathogenicity of influenza virus. To do so, we initially monitored the cytokine response after viral infection in vitro and in vivo. Infection of the RG-AIV-NS3 virus in both A549 and DF1 cells stimulated a higher level of interferon-β (IFN-β) and TNF-α than the other two viruses. This effect seemed to be dependent on the host cell, as a higher level of IFN-β was detected in M1 cells infected by RG-AIV-T375G virus (). Notably, expression of Mx, the IFN stimulated gene (ISG), was increased by about 2-fold in DF1 cells infected with RG-AIV-NS3, as compared with the other two viruses.
Figure 6. Expression profiles of cytokines in mammalian or avian cells infected with reassortant AIVs. Human A549 (A), murine M1(B), and chicken DF1 (C) cells were infected with reassortant AIVs at MOI of 0.1. Total RNA was extracted at 6 hpi. The expression levels of cytokines, including IFN-β, MX and TNF-α were detected by qRT–PCR. The expression level of each cytokine was initially normalized to that of mock infection. All experiments were conducted in triplicate. *p < 0.05 comparing the particular virus with others.
Consistent with the findings in vitro, RG-AIV-NS3 elicited a robust cytokine response in mice (). At 12 hpi, mice infected with RG-AIV-NS3 had significantly higher mRNA and protein levels of IFN-β than those infected with the other two viruses ((A,B)). Moreover, RG-AIV-NS3 stimulated higher TNF-α expression in mice than RG-AIV-WT ((C)). It appeared that high cytokine expression was correlated with viral replication efficiency, since significantly higher M gene transcript levels were detected in mice infected by RG-AIV-NS3 compared to the other viruses. In contrast to these early differences, the distinct pattern of RG-AIV-NS3 response was not apparent at 24 hpi.
Figure 7. Expression profiles of cytokines and viral RNA in mice infected with reassortant AIVs. Mice (n = 5) were infected with reassortant AIVs, and blood were collected at 12 and 24 hpi. The expression levels of cytokines, including IFN-β and TNF-α were detected by qRT–PCR (A, C), or ELISA (B). Moreover, at 24 hpi, blood was collected and mice were sacrificed. IFN-β protein level in blood was detected by ELISA (B), and viral RNA (M gene) in lung tissue was measured (D). All experiments were conducted in triplicate. Significant differences between two groups are denoted by different letters.
Ultimately, the survival rates of mice infected with the three viruses were evaluated. As shown in (A), each virus caused continuous weight loss in mice up to 9 days post-infection (dpi), while mice that survived RG-AIV-T375G infection began to regain weight from 10 dpi onward. Notably, none of the mice receiving RG-AIV-WT or RG-AIV-NS3 survived to 10 dpi ((B)). The survival rate of mice infected by RG-AIV-T375G virus was significantly higher than the survival rates of mice infected with RG-AIV-WT or RG-AIV-NS3 ((C)). Moreover, the mean survival time of mice infected by RG-AIV-T375G virus was 12.3 days, which was longer than the mean survival times after RG-AIV-WT (5 days) or RG-AIV-NS3 (5.3 days) infection. Along with the cytokine expression and M gene expression results, these findings suggest that influenza virus expressing only full length NS1 is highly attenuated in the mouse model.
Figure 8. Survival rates of mice infected with reassortant AIVs. Mice (n = 7) were infected with reassortant AIVs. Infected (WT, NS3, or T375G virus) and control mice (PBS) were monitored for 14 days post-infection (dpi) for body weight loss (A) and survival rate (B and C). ***p < 0.005; ns, not significant (p > 0.05).
Discussion
In the current study, we found that NS1 RNA from clade 2.3.4.4 H5N2 HPAIV (strain 1031) encodes two proteins, full-length NS1 and NS3. To examine the function of NS3, we generated virus bearing wild-type NS1 (RG-AIV-WT) and two reassortant viruses, RG-AIV-T375G and RG-AIV-NS3. Using these tools, we uncovered the comparative functions of full-length NS1 (herein called T375G) and NS3 in the context of influenza virus infection. We found that the two proteins play distinct host-dependent roles in determining the efficiency of infection. Expression of T375G conferred a growth advantage in the chicken cell line, while NS3 expression caused the virus to replicate more efficiently in three types of mammalian cells. Moreover, the NS3 protein is not redundant with NS1 in terms of its molecular action. Our in vitro studies showed that NS3 enhances viral RdRp activity and suppresses PKR activation to a greater extent than full-length NS1 alone or both proteins expressed from the wild-type NS1 gene. In the mouse model, RG-AIV-T375G was highly attenuated, while RG-AIV-NS3 exhibited similar virulence to RG-AIV-WT.
Influenza A virus mutates frequently, and NS1 is especially prone to adaptive mutations [Citation27]. Previous work has shown that NS1 RNA can yield various proteins by different mechanisms. Here, we show that the NS1 RNA of H5N2 (strain NS1031) encodes an additional NS3 protein, which is translated from an alternatively spliced NS1 transcript. In the NS1 gene of H5N2, a novel splice donor dinucleotide (GT375) is constituted in the 125G codon, leading to the expression of a shortened NS1 isoform with internal deletion of residues 126–168 from the spliced NS3 transcript ( and Supplementary Figure S2).
It is well documented that mutations can give rise to diverse activities of influenza viral proteins that are critical for pathogenesis in new host species [Citation28]. In line with this observation, the presence of the NS3 transcript has been reported in human H3N2, and it was shown to facilitate adaptation in mice models [Citation28]. Initially, the D125G mutation in NS1 was thought to be one of two residues responsible for the increased virulence of human A/Aichi/2/68 (H3N2) virus in BALB/c mice during blind passaging [Citation28]. Similarly, an independent study demonstrated that 11 individual substitutions in the NS1 gene were selected during serial lung-to-lung passaging of human A/HK/1/1968 (H3N2) in CD-1 mice [Citation29]. Among those mutations, the A374G substitution (GAT→GGT) causes a D125G missense mutation in the full-length NS1. In addition, the A374G substitution introduces a novel donor splice site in the NS gene, which ultimately gives rise to a novel influenza A viral protein, NS3. As compared with parental H3N2 virus bearing wild-type NS, strains with the A374G substitution exhibit enhanced replication and virulence of mouse-adapted (MA) D125G virus in the mouse model. Furthermore, the other adaptive mutations were also individually characterized [Citation30,Citation31]. In that set of studies, MA D125G virus expressing both NS1 and NS3 had gain-of-function properties and no apparent loss of function, except for a lack of binding to CPSF30 (30-kDa cleavage and polyadenylation specificity factor) [Citation30].
Similar to these findings regarding human influenza H3N2, our study revealed that the NS gene of the H5N2 virus strain 1031 also encodes multiple proteins. Sequence alignment of the NS1 genes from several H5N2 strains isolated in Taiwan between 2013 and 2015 revealed that the 1031 strain (but not other LPAIVs) harbors the T375 nucleotide [Citation21]. Similar to A374G substitution in MA H3N2, T375 in the H5N2 NS1 gene also comprises a GU splice donor sequence. Hence, the gene encodes both the full-length NS1 and NS3, which is translated from the spliced transcript and carries an internal deletion ((A)). The H5N2 NS3 protein appears to be exclusively localized in the nucleus and exhibits a punctate pattern upon staining ((C)), which is consistent with the D125G variant of MA H3N2 virus [Citation31]. As shown in , expression of NS3 is relatively lower than that of NS1 in M1 cells infected by RG-AIV-WT at 12 hpi. However, the lower level of NS3 is probably not due to inefficient splicing in M1 cells, since the ratio of NS3/NS1 mRNA showed no differences among the three tested mammalian cell types ((C)). Whether the lower level is due to a shorter half-life of NS3 in M1 cells will require further investigation.
Current understanding of NS3 is mainly based on characterization of MA D125G H3N2 virus, which expresses both NS1 and NS3 proteins. Hence, we wanted to clarify the comparative functions of NS3 and full-length NS1 individually. Surprisingly, among the three reassortant viruses we generated for this study, RG-AIV-NS3 had the highest multiplication kinetics in three mammalian cell lines but not in chicken DF1 cells. In contrast, RG-AIV-T375G (expressing full-length NS1 only) replicated poorly in mammalian cells, but it had a growth advantage in chicken DF1 cells (). It is therefore likely that an influenza virus (e.g. RG-AIV-WT) bearing an NS gene that expresses both NS1 and NS3 could transmit swiftly between mammalian and avian species. Of note, the NS segment of strain 1031 was quickly replaced by one from other strains in Taiwanese poultry farms. This observation, along with evidence from a previous study [Citation21], leads us to propose that the NS segment of strain 1031 may not be compatible with other inner genes (particularly those encoding the RdRp complex) derived from other lineages of H5 avian influenza virus. For the current study, we attempted to generate a reassortant virus expressing NS3 or T375G only on the genetic background of H6N1 avian influenza, as the system has been readily used for generating AIVs to express the wild-type NS segment of strain 1031 [Citation21]. However, we were unfortunately not able to create a stable version of the virus. Therefore, we postulate that the coexistence of full-length NS1 protein and NS3 could possibly compensate for incompatibility of NS3 (or T375G) and inner genes derived from avian influenza (i.e. H6N1). As shown in (C), despite the efficient production of RG-AIV-NS3 in mammalian cells, the virus replicated poorly in DF1 cells. Meanwhile, T375G (full-length NS1) had the highest yield among the three viruses in DF1 cells, revealing the remarkable influence of NS1 and NS3 on overall infection efficiency. Since RG-AIV-NS3 could be successfully rescued on the genetic background of human H1N1 subtype and NS3 protein had an enhanced effect on H1N1 RdRp activity ((A)), we strongly suspect that reassortment of the NS segment from the 1031 strain into human influenza viruses could enhance the fitness and increase the likelihood of interspecies transmission. As evidenced in the mouse experiment (), viruses bearing WT NS1031 or NS3 alone is more virulent than the RG-AIV-T375G that further strengthens the hypothesis that existence of NS3 increases the overall replication efficiency and also pathogenicity of influenza in mice.
NS1 is expressed as early as 4 hpi [Citation32,Citation33], and the protein functions by several mechanisms to confer a growth advantage to influenza virus at early infection times. Apart from its well-known dsRNA binding activities, NS1 also promotes viral genome replication and hijacks cellular protein translation through respective interactions with viral RdRp [Citation34] and cellular factor, eIF4G I [Citation35]. Moreover, NS1 binds to several cellular partners via its ED domain to attenuate the innate immune response [Citation17]. NS3 lacks residues 126–168 located in the ED of NS1 (Supplementary Figure S2). Intriguingly, NS3 retains the known NS1 function of counteracting PKR activation ((A)), and this action is probably at least partly due to the strong interaction between NS3 and PKR ((A)). Regions of NS1 contributing to regulation of PKR activity have been extensively studied. Two independent studies revealed that residues 123–127 in ED [Citation18], or two arginine residues (35 and 46) near the N-terminus of NS1 are essential for binding to and blocking activation of PKR [Citation36]. Sequence alignment indicated the two arginine residues are conserved in NS3, whereas residues 126 and 127 are missing (Supplementary Figure S2). Nevertheless, it seems deletion of residues 126–127 does not affect the inhibitory effect of NS3 on PKR activation. Of note, the importance of residues 126–127 on suppression of PKR inactivation was only documented for H3N2 (Udorn strain) [Citation18], and the effect was not shown for NS1 of H1N1 (strain PR8) [Citation36]. Similarly, the effects of the two-N-truncated NS1 isoforms encoded from Udorn strain and PR8 strain on IRF-3 activation were inconsistent [Citation11]. It was speculated that the apparent discrepancy could be simply due to the different modes of action for NS1 on PKR inhibition, or it could be due to structural heterogeneity and conformational plasticity of NS1 proteins between strains [Citation37]. In its role as a dsRNA-binding protein, NS1 exerts versatile activities and dynamic interactions. It can form dimers or oligomers with various cellular counterparts via ED-ED “helix-helix” interactions. One might expect that the sequence variations among the NS1 homologues could reflect different secondary and tertiary structures that would present distinct available functional spaces and interfaces for the formation of higher-order structures with PKR. Hence, the structure of each NS1-interactor complex could be unique and should be interpreted at a strain-specific level.
In conclusion, this study reveals that the NS segment from the HPAIV H5N2 strain 1031 encodes both NS1 and NS3 proteins. The data herein not only define the expression mechanism of NS3 but also clarify the function of NS3 using in vitro and in vivo models. Despite the internal deletion of residues 126-168, NS3 exhibits conservation of essential NS1 functions, especially the suppression of PKR activation and enhancement of viral RdRp. Moreover, the mouse model experiments confirm that NS3 is a key virulence determinant, with T375G protein rendering the virus remarkably attenuated. Since the full length NS1 and NS3 proteins differentially influence viral growth in avian and mammalian cells, reassortant viruses harboring G374 and T375 within the NS segment likely possess a high potential for interspecies transmission. Special attention should be paid to this sequence signature when monitoring novel and emerging viral species or antigenic variants.
Supplemental Material
Download Zip (9.2 MB)Acknowledgements
The authors thank Professor M.S. Chang in Department of Biochemistry and Molecular Biology, in National Cheng Kung University, Taiwan, and also Professor Bernard Moss in NIAID, NIH, USA for providing M1 cells and PKR-KO A549 cells, respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhang Z, Chen D, Chen Y, et al. Spatio-temporal data comparisons for global highly pathogenic avian influenza (HPAI) H5N1 outbreaks. PLoS One. 2010 Dec 20;5(12):e15314.
- Lee DH, Bertran K, Kwon JH, et al. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J Vet Sci. 2017 Aug 31;18(S1):269–280.
- Lu YS S, Shieh T, Lee HK, et al. Isolation and identification of an influenza A virus in ducks in Taiwan. J Chin Soc Veter Sci. 1985;11:23–34.
- Lee MS, Chang PC, Shien JH, et al. Genetic and pathogenic characterization of H6N1 avian influenza viruses isolated in Taiwan between 1972 and 2005. Avian Dis. 2006 Dec;50(4):561–571.
- Cheng MC, Soda K, Lee MS, et al. Isolation and characterization of potentially pathogenic H5N2 influenza virus from a chicken in Taiwan in 2008. Avian Dis. 2010 Jun;54(2):885–893.
- Lee CC, Zhu H, Huang PY, et al. Emergence and evolution of avian H5N2 influenza viruses in chickens in Taiwan. J Virol. 2014 May;88(10):5677–5686.
- Lee MS, Chen LH, Chen YP, et al. Highly pathogenic avian influenza viruses H5N2, H5N3, and H5N8 in Taiwan in 2015. Veter Microbiol. 2016 May 1;187:50–57.
- Chen LH, Lee DH, Liu YP, et al. Reassortant Clade 2.3.4.4 of highly pathogenic Avian Influenza A(H5N6) Virus, Taiwan, 2017. Emerg Infect Dis. 2018 Jun;24(6):1147–1149.
- Krammer F, Smith GJD, Fouchier RAM, et al. Influenza. Nat Rev Dis Primers. 2018 Jun 28;4(1):3.
- Wise HM, Barbezange C, Jagger BW, et al. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res. 2011 Sep 1;39(17):7775–7790.
- Kuo RL, Li LH, Lin SJ, et al. Role of N terminus-truncated NS1 proteins of influenza A virus in inhibiting IRF3 activation. J Virol. 2016 May;90(9):4696–4705.
- Qian XY, Alonso-Caplen F, Krug RM. Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J Virol. 1994 Apr;68(4):2433–2441.
- Ji ZX, Wang XQ, Liu XF. NS1: a Key protein in the “game” between influenza A virus and host in innate immunity. Front Cell Infect Microbiol. 2021;11:670177.
- Chow KT, Gale M, Jr., Loo YM. RIG-I and other RNA sensors in antiviral immunity. Annu Rev Immunol. 2018 Apr 26;36:667–694.
- Jureka AS, Kleinpeter AB, Tipper JL, et al. The influenza NS1 protein modulates RIG-I activation via a strain-specific direct interaction with the second CARD of RIG-I. J Biol Chem. 2020 Jan 24;295(4):1153–1164.
- Mibayashi M, Martinez-Sobrido L, Loo YM, et al. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007 Jan;81(2):514–524.
- Ayllon J, Garcia-Sastre A. The NS1 protein: a multitasking virulence factor. Curr Topics Microbiol Immunol. 2015;386:73–107.
- Min JY, Li S, Sen GC, et al. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007 Jun 20;363(1):236–243.
- Gack MU, Albrecht RA, Urano T, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009 May 8;5(5):439–449.
- Twu KY, Noah DL, Rao P, et al. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J Virol. 2006 Apr;80(8):3957–3965.
- Wang WC, Kuan CY, Tseng YJ, et al. The impacts of reassortant Avian Influenza H5N2 Virus NS1 proteins on viral compatibility and regulation of immune responses. Front Microbiol. 2020;11:280.
- Martinez-Sobrido L, Garcia-Sastre A. Generation of recombinant influenza virus from plasmid DNA. J Vis Exp. 2010 Aug 3;42.
- Tseng YY, Lin FY, Cheng SF, et al. Functional analysis of the short isoform of orf virus protein OV20.0. J Virol. 2015 May;89(9):4966–4979.
- Massin P, Rodrigues P, Marasescu M, et al. Cloning of the chicken RNA polymerase I promoter and use for reverse genetics of influenza A viruses in avian cells. J Virol. 2005 Nov;79(21):13811–13816.
- Spackman E, Suarez DL. Type A influenza virus detection and quantitation by real-time RT-PCR. Methods Mol Biol. 2008;436:19–26.
- Liu R, Moss B. Opposing roles of double-stranded RNA effector pathways and viral defense proteins revealed with CRISPR-Cas9 knockout cell lines and vaccinia virus mutants. J Virol. 2016 Sep 1;90(17):7864–7879.
- White MC, Lowen AC. Implications of segment mismatch for influenza A virus evolution. J Gen Virol. 2018 Jan;99(1):3–16.
- Narasaraju T, Sim MK, Ng HH, et al. Adaptation of human influenza H3N2 virus in a mouse pneumonitis model: insights into viral virulence, tissue tropism and host pathogenesis. Microbes Infect. 2009 Jan;11(1):2–11.
- Selman M, Dankar SK, Forbes NE, et al. Adaptive mutation in influenza A virus non-structural gene is linked to host switching and induces a novel protein by alternative splicing. Emerg Microb Infect. 2012 Nov;1(11):e42.
- Forbes NE, Ping J, Dankar SK, et al. Multifunctional adaptive NS1 mutations are selected upon human influenza virus evolution in the mouse. PLoS One. 2012;7(2):e31839.
- Forbes N, Selman M, Pelchat M, et al. Identification of adaptive mutations in the influenza A virus non-structural 1 gene that increase cytoplasmic localization and differentially regulate host gene expression. PLoS One. 2013;8(12):e84673.
- Tseng YY, Kuan CY, Mibayashi M, et al. Interaction between NS1 and cellular MAVS contributes to NS1 mitochondria targeting. Viruses. 2021 Sep 23;13(10).
- Tsai CF, Lin HY, Hsu WL, et al. The novel mitochondria localization of influenza A virus NS1 visualized by FlAsH labeling. FEBS Open Bio. 2017 Dec;7(12):1960–1971.
- Marion RM, Zurcher T, de la Luna S, et al. Influenza virus NS1 protein interacts with viral transcription-replication complexes in vivo. J Gen Virol. 1997 Oct;78(Pt 10):2447–2451.
- Aragon T, de la Luna S, Novoa I, et al. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol Cell Biol. 2000 Sep;20(17):6259–6268.
- Schierhorn KL, Jolmes F, Bespalowa J, et al. Influenza A virus virulence depends on Two amino acids in the N-terminal domain of Its NS1 protein To facilitate inhibition of the RNA-dependent protein kinase PKR. J Virol. 2017 May 15;91(10).
- Hale BG. Conformational plasticity of the influenza A virus NS1 protein. J Gen Virol. 2014 Oct;95(Pt 10):2099–2105.