
ABSTRACT
Avian influenza H5N1 is a highly pathogenic virus that primarily affects birds. However, it can also infect other animal species, including mammals. We report the infection of nine juvenile red foxes (Vulpes vulpes) with Highly Pathogenic Avian Influenza A type H5N1 (Clade 2.3.4.4b) in the spring of 2022 in the central, western, and northern regions of New York, USA. The foxes displayed neurologic signs, and examination of brain and lung tissue revealed lesions, with brain lesions ranging from moderate to severe meningoencephalitis. Analysis of tissue tropism using RT-PCR methods showed a comparatively lower Ct value in the brain, which was confirmed by in situ hybridization targeting Influenza A RNA. The viral RNA labelling was highly clustered and overlapped the brain lesions, observed in neurons, and grey matter. Whole viral genome sequences obtained from the affected foxes were subjected to phylogenetic and mutation analysis to determine influenza A clade, host specificity, and potential occurrence of viral reassortment. Infections in red foxes likely occurred due to preying on infected wild birds and are unlikely due to transmission between foxes or other mammals.
Introduction
Influenza A virus jumps species barriers and adapts to its host(s) through changes in the receptor binding and fusion envelope glycoproteins hemagglutinin (HA) and neuraminidase (NA). The cleavability of the HA is an essential determinant of the virulence of avian influenza (AI) viruses [Citation1,Citation2]. Spillover of highly pathogenic AI virus (HPAIV) from wild to domestic birds was first observed in 1996 with the virus A/goose/Guangdong/1/1996. This virus has caused repetitive epizootics in poultry and wild birds since 2015. By late 2020, the clade 2.3.4.4b H5N1 subtype emerged as a new variant and became predominant in outbreaks detected worldwide [Citation3]. The current Eurasian strain of the HPAI H5N1 epizootic was first detected in North America in late 2021 in Canada. Extensive wild bird surveillance efforts by the U.S. Department of Agriculture (USDA), Animal and Plant Health Inspection Service (APHIS) National Wildlife Disease Program, National Animal Health Laboratory Network (NAHLN), and National Veterinary Services Laboratories (NVSL) determined that H5N1 clade 2.3.4.4b was detected in wild waterfowl in two states in the Atlantic flyway, North Carolina and South Carolina, in January 2021 [Citation4]. H5N1 HPAI has subsequently been confirmed in wild birds, backyard flocks, commercial facilities, and wild mammals in the US [Citation5]. Carnivorous animals, including red foxes (Vulpes vulpes), are at high risk of infection for HPAI H5N1 due to their behaviour and habit of preying on wild birds or scavenging carcasses [Citation6].
In late 2020, a report from the United Kingdom in a wildlife rehabilitation facility described the detection of HPAIV infection in red foxes associated with infected swans quarantined nearby [Citation7]. In 2021, HPAIV Eurasian lineage H5 clade 2.3.4.4.b was detected in two fox cubs in the Netherlands with signs of respiratory and neurological disease. Viral sequences from these two foxes clustered with other sequences recovered nearby from HPAIV-infected waterfowl and birds of prey [Citation8]. In the winter and spring of 2021–2022, HPAIV was confirmed in an Ezo red fox (Vulpes vulpes schrenki) and a tanuki (Nyctereutes procyonoides albus) found in a garden [Citation9].
Unlike typical influenza pathotypes, studies have shown that HPAI H5N1 spreads directly from the olfactory mucosa to the central nervous system, causing severe neurologic manifestations such as encephalitis and meningitis [Citation10]. In this study, we have identified and characterized HPAI H5N1 clade 2.3.4.4b in juvenile red foxes (Vulpes vulpes) from New York State (NYS) collected in the spring of 2022. The fox kits exhibited neurologic signs, including tremors, blindness, and lethargy before death. Neuronal infection by HPAIV was confirmed in red foxes by in situ hybridization.
Materials and methods
Specimen collection
Between April and May 2022, 29 juvenile red foxes (Vulpes vulpes) (1 month to 1-year-old) were submitted to the Animal Health Diagnostic Center (AHDC) at Cornell University for necropsy or ancillary testing. At antemortem examination, many of the fox kits were observed in extreme distress, with respiratory and neurologic signs including seizures. Many animals were humanely euthanized due to their poor condition ().
Table 1. Sample intake metadata for foxes with Influenza A diagnostic RT-PCR cycle threshold (Ct ≤ 45) values.
When possible, complete post-mortem examinations were performed on the affected foxes. Standard procedures performed included gross and histologic evaluations. Additionally, frozen tissues were collected at necropsy and subjected to influenza virus RT-PCR. In some cases, paraffin embedded formalin fixed (FFPE) tissues were subjected to RT-PCR.
Nucleic acid extraction and influenza A RT-PCR
To investigate the potential involvement of influenza A in the disease presentation in foxes, tissue samples (lung, brain, pooled tissue) and respiratory (oropharyngeal) swabs were subjected to nucleic acid extraction using the standard AI protocol recommended by the National Animal Health Laboratory Network (NAHLN) with slight modification [Citation12]. Modifications included the use of MagMax™ Clarifying Solution (Cat # A46972C) and a mechanical lysis step [Citation11,Citation13]. RT-PCR was performed either on an ABI 7500 or on the Quant Studio 12 K Flex OpenArray (OA) (ThermoFisher Scientific) [Citation11,Citation13]. Once it was determined that these samples were positive for AI-Matrix PCR, the virus was subtyped using the H5 RT-PCR NAHLN assay [Citation12]. Of the 29 fox kits received between April and May 2022, 24 were tested for influenza A; ten were found positive for AI-Matrix PCR and “non-negative” (presumptive positive) for AI-PCR (NAHLN) and H5PCR (NVSL) [Citation12]. Notably, the ten foxes that tested positive for influenza PCR in the present study were also submitted to the NYS Department of Health for both rabies fluorescent antibody testing and canine distemper PCR, with negative results.
Tissue tropism
To assess tissue distribution of HPAI in the affected fox kits, we collected brain, heart, lung, kidney, liver, spleen, mesenteric lymph node, and distal small intestine from six juvenile male foxes. As described above, these tissues were subjected to nucleic acid extraction and RT-PCR for the AI-Matrix gene. RT-PCR Ct data were statistically analysed by performing a two-way analysis of variance (ANOVA) in GraphPad Prism v.9.5.1.
RNA in situ hybridization (RNA-ISH) was performed on the resulting positive samples for three specimens following the manufacturer’s instructions for the RNAscope™ 2.5 HD Assay-Red (Advanced Cell Diagnostics, Inc.) for single RNA molecular detection and visualization. The V-InfluenzaA-H5N8-M2M1-C1 (cat. no. 1048221-C1) probe was used to test the samples. RNAscope™ Probe – Cl-UBC Positive Control (cat. no. 409851), and RNAScope probe DapB (cat. no. 310043) were used as positive and negative controls [Citation14].
Whole genome sequencing methods
Of the ten influenza-positive foxes, nine underwent whole genome sequencing. One formalin-fixed, paraffin-embedded tissue sample was not included since it had a high Ct value (Ct = 38.132) and was unlikely to generate quality sequences (see sample metadata in ). All nine samples were sequenced using Oxford Nanopore Technologies (ONT, Mk1C, MinION R9.4.1) methods, and five also were sequenced using the Illumina iSeq 100 platform (I1 Reagent V2, 2×150). Frozen brain and lung tissue were the preferred sample type for sequencing. The purified nucleic acid was subjected to multi-segment reverse-transcription PCR (M-RT-PCR) for whole genome amplification, a method that takes advantage of highly conserved regions at the ends of the 8 RNA segments of IA viruses [Citation15,Citation16]. Illumina libraries were prepared using the Illumina DNA prep and Oxford Nanopore (ONT) libraries were prepared using the EXP-NBD196 and SQK-LSK109 kits [Citation17].
Sequencing analysis and phylogenetic analysis methods
The fast5 files for each of the nine samples run using ONT sequencing methods were demultiplexed and trimmed using Guppy v.6.4.6 [Citation18]. Canidae host reads were removed using NanoLyse v.1.2.0 and filtered using NanoFilt V.2.8.0 from the fastq files [Citation19]. Reads were classified using Kraken2 v.2.1.0 and Kaiju 1.9.2 [Citation20,Citation21]. Fastq reads were aligned to AI H5N1 sequence (A/Thailand/1(KAN-1)/2004 (H5N1)) (NCBI AY626144-51) as the reference using Minialign v.0.4.4 [Citation22]. Consensus sequences were generated using BCFtools v.1.14 and polished using Medaka v.1.4.3 with ONT read files or with both ONT and Illumina read files when available [Citation23]. Consensus sequences were published on Global Initiative on Sharing All Influenza Data (GISAID) and NCBI (Supplemental Table 1).
To assess potential reassortment, we produced a genotyping database of the segment sequences described as generating the original H5N1 index virus A/Eurasian wigeon/Netherlands/1/2020 by Cui et al. 2022 and Shi et al. 2023. Included in this database was a recently detected avian H5N1 sequence in NYS and the reference sequence used to in the reference-based alignment method (Supplemental Table 2) [Citation24,Citation25]. After reads were filtered and host Canidae reads removed, alignments were run against this database using minimap2, and resulting coverage and read depth statistics were assessed using Samtools v.1.15.1 [Citation26,Citation27].
Whole genome mutation analysis and interpretation were performed using FluSurver through GISAID EpiFlu, and the classical H3N2 HA mutation nomenclature was presented [Citation28]. Additional mutation analysis was performed for critical amino acid substitutions that were not included in the output from FluSurver, by performing protein sequence alignments in Geneious Prime v.2022.1.1. Microreact was used to visualize data by uploading metadata and a phylogenetic tree Newick file made in Geneious Prime v.2022.1.1, Tamura-Nei genetic distance model, and Neighbor-joining consensus tree built from the HA consensus sequences [Citation29,Citation30].
Segment 4, hemagglutinin (HA) assemblies were initially analysed using the H5 Clade Classification tool available on the Influenza Research Database (IRD) [Citation31]. Avian H5N1 HA sequences and clade annotations were downloaded from IRD (sequences before 2020) and GISAID (2020-recent). We performed phylogenetic analysis by constructing alignments with published genomes in GenBank, IRD, and GISAID using MAFFT (v7.453). Phylogenetic trees were created using the Tamura-Nei model and bootstrap analysis (n = 1000) in Mega (v.11.0.9), collapsed and annotated branches using FigTree (v1.4.3.exe) [Citation32–35].
Results and discussion
Case history
Twenty-nine foxes were brought to the Cornell University Animal Health Diagnostic Center (AHDC) between April and May 2022, and 24 red foxes were submitted for necropsy from the NYS Department of Environmental Conservation (DEC) Wildlife Health Program or the Janet Swanson Wildlife Hospital at Cornell. The NYS DEC necropsied five foxes, and tissues were sent to the AHDC for ancillary testing. Antemortem, many fox kits were observed in extreme stress and pain. Many exhibited respiratory and neurologic signs, including seizures, while some had been hit by cars or found severely injured. Of the 29 foxes, 24 were tested for Influenza A virus due to the clinical signs presented at intake, and ten of those 24 (10/24) tested positive for Influenza A matrix. In , we present a summary of the clinical history and other relevant information including collection data, geographic location, reported signs, and resulting diagnostic Ct value on the 10 foxes that tested positive for influenza A.
Pathological evaluations
Five of the 10 HPAI positive animals (4 m, 1 F) were subjected to post-mortem examinations. Foxes ranged from approximately 1–5 months old, with body condition scores noted from poor to good. Gross pulmonary lesions spanning edema to diffuse consolidation affected all foxes. The brain exhibited no gross abnormalities, though one animal displayed slight calvarial doming.
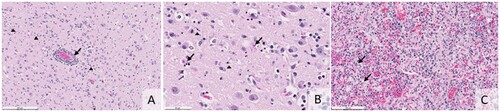
Histologically, all five animals displayed lesions in the brain and lung. Brain lesions ranged from moderate to severe meningoencephalitis characterized by an influx of lymphocytes, plasma cells, macrophages, rarer neutrophils, and reactive glial cells. Variably sized areas of necrosis stippled the cerebral cortex (A and B). The most prominent changes affected the grey matter, although white matter lesions were also present. Four animals exhibited mild to severe necrotizing interstitial pneumonia with fibrin, neutrophils, and occasional type II pneumocyte hyperplasia (C). The fifth animal’s lungs were mildly edematous, with some histocytes in the alveoli. Other lesions were interpreted as unrelated to the influenza infection. These included multifocal mineralization with myofiber degenerate (n = 2) and multifocal hepatic necrosis (n = 1). One animal had pyelitis and nephritis consistent with an ascending bacterial infection. However, this lesion was significant, meningoencephalitis was interpreted as a malignant lesion. Unlike the other animals, this fox was in poor body condition.
Figure 1. (A) Histologic section shows monocular perivascular cuffing with rarer neutrophils (arrow). Cellular debris and reactive glia are present throughout the grey matter (arrowheads, H&E, bar = 200 microns). (B) Histology of the cerebral cortex illustrating neuronal necrosis (arrows) and small deposits of nuclear debris in the grey matter (arrowheads, H&E, bar = 50 microns). (C) Histology of the lung shows macrophages, neutrophils, and fibrin flooding and obliterating the alveoli. In areas, there is type 2 pneumocyte hyperplasia (arrows, H&E, bar = 100 microns).
Host tissue tropism
RT-PCR results
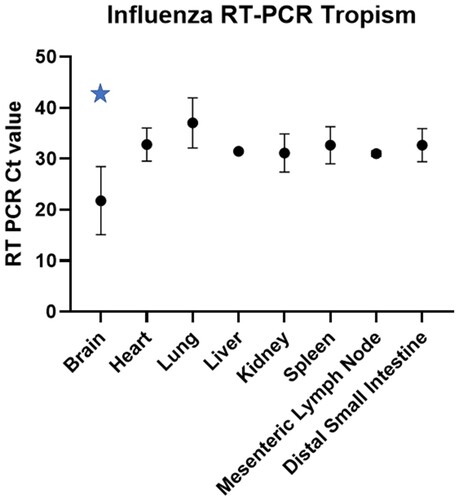
Initial viral tropism was assessed by RT-PCR in brain, heart, lung, liver, kidney, spleen, mesenteric lymph node and distal small intestine. Viral RNA loads as determined by RT-PCR Ct values indicated statistically lower viral loads in the brain when compared to all other tissues evaluated using a two-way ANOVA (p = 0.0001) (, data in Supplemental Table 3).
Figure 2. RT-PCR Ct values (column mean, error bars) for each IA matrix positive fox (n = 4) tissue included in the study. The blue star indicates statistically lower Ct values (p = 0.0001) in the brain when compared to the other tissue using two-way ANOVA.
In situ hybridization
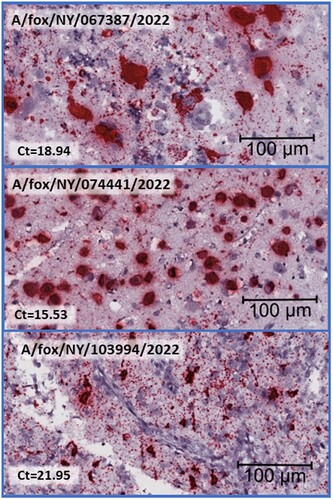
The most intense and consistent staining is in the brain, with a strong viral labelling detected within histological lesions. Staining is confined to the grey matter, and most of the cells that exhibit staining are neurons. Large amounts of viral RNA fill the cytoplasm and extend into the neuronal process. Stippling of staining is present in the adjacent neuropils and is presumed to be within the axons ().
Figure 3. Histological brain sections subjected to Influenza A RNA-ISH at high magnification, size bar at 100 µm, and corresponding Ct value from the Influenza matrix RT-PCR is included. Clustered chromogenic staining indicates highly concentrated RNA signalling in brain lesions.
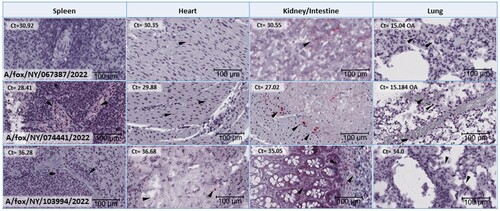
Less intense staining is present in other organs, including the lungs, kidneys, and heart. Of these, staining and lesions are most consistently observed in the lungs. Less intense, labelling was present in the cytoplasm of macrophages and possibly Type II pneumocytes within the areas of inflammation. Similarly, faint labelling in the kidney and the heart is limited to the areas of inflammation and appear to be restricted to macrophages primarily ().
Figure 4. Histological tissue sections subjected to Influenza A RNA-ISH for spleen, heart, kidney (for A/fox/NY/067387/2022 and A/fox/NY/074441/2022) or intestine (A/fox/NY/103994/2022) and lung. Each image is at the same magnification and has a size bar at 100 µm in the lower right-hand corner. Arrows indicate chromogenic viral RNA labelling that may be difficult to locate due to a lower level of virus presence than in brain tissue sections. No staining was observed for A/fox/NY/067387/2022 spleen tissue section. Corresponding RT-PCR Ct values on each of the tissues depicted are included in the upper left-hand corner for each image.
Phylogenetic and mutation analysis
Influenza viruses evolve quickly through the accumulation of mutations in their genomes and/or reassortment of genomic sequences between strains. The genome of IA viruses comprises eight segments of negative-sense, single-stranded RNA organized as individual ribonucleoprotein (RNP) complexes. The novel H5N1, A/Eurasian wigeon/Netherlands/1/2020(H5N1), was first detected in the Netherlands in October 2020. It was a descendant of the reassortment of an H5N8 virus (providing the HA and M genes) with five other influenza viruses of different subtypes. Influenza viruses of the same clade may have different NA and internal genes and be classified into different genotypes [Citation24,Citation25].
Phylogenetic analysis of the HA segment of HPAIV obtained from foxes combined with spatiotemporal analyses showed that foxes collected on the same day and location had genetically identical HA sequences, two of which were found to be of the same genotype ().
Figure 5. Spatiotemporal spread of H5N1 viruses in foxes. (A) Map the location where each animal was initially found and provide the geographic relation within Upstate NY. The circle size indicates the number of foxes found in that location. (B) Phylogenetic analysis of the HA segment showed that samples found in location contained Influenza strains that were genetically related. (C) Temporal timeline indicating when each fox had been found or collected between April and May 2022, including the genotypic relationship for each of the seven samples that yield 100 percent coverage of each of the eight segments. (D) Two genotypes of Influenza A H5N1 that were found in this study. The colour of the bar indicates the closest donor strain of the gene segment, which can be found in the “Segment Origin” table. The IA virus coloured blue is referred to as genotype 1, and red is genotype 2.
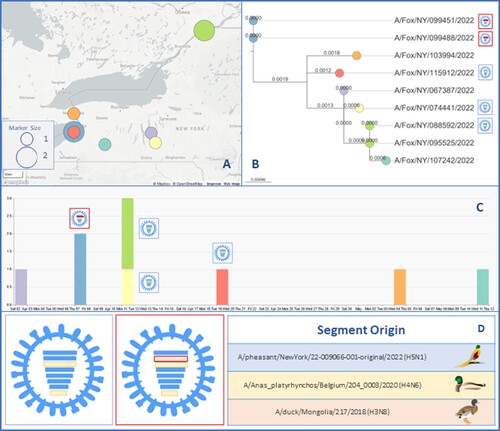
The nine H5N1 viruses detected in red foxes in this study formed two distinct genotypes. HPAIV sequencing reads for all of the foxes aligned to the segments of A/pheasant/NewYork/22-009066-001-original/2022(H5N1) for polymerase subunit basic 2 (PB2), hemagglutinin (HA), nucleocapsid (NP), neuraminidase (NA), and matrix protein (M) and with A/Anas_platyrhynchos/Belgium/204_0003/2020(H4N6) for polymerase subunit acidic (PA) and non-structural protein (NS), when available. The two genotypes (G1 and G2) were identified based on the origin of the polymerase subunit basic 1 (PB1) segment, with three of the foxes having PB1 descend from A/pheasant/NewYork/22-009066-001-original/2022(H5N1) and two from A/duck/Mongolia/217/2018(H3N8). shows ribonucleoprotein (RNP) diagrams with coloured bars indicating the segment origin strain and G1 outlined in blue and G2 in red. Four of the samples did not have full sequence coverage for PB1 segments (due to a low viral loads) and thus were not genotyped.
All available H5 HA sequences (as of March 2023) derived from mammals worldwide from 2021 to 2023 were downloaded and subjected to phylogenetic analysis. The sequences originated from three continents, North America (123, 69.9%), Europe (51, 29.0%), and Asia (2, 1.1%) (A). There were 176 mammalian influenza virus sequences published worldwide, from Red fox (109, 61.9%), Striped skunk (21, 11.9), Harbor seal (13, 7.4%), European polecat (6, 3.4%), Mink (6, 3.4%), Virginia opossum (2, 1.1%), Raccoon (2, 1.1%), Ferret (2, 1.1%), along with Wild mink, Tanuki, Otter, Grey seal, fisher, ferret, Feline, Ezo red fox, Coyote, Bush dog, Bobcat, bear, and badger (all 0.6%).
Figure 6. (A) A stacked column chart presenting the number of mammalian HPAI H5N1 sequences worldwide and published on GISAID between 2021 and 2023. The x-axis consists of individual countries in Europe, states in the US, provinces in CA, and Japan. (B) Mutation analysis summary for each of the eight genome segments. Previously reported effects of such mutations, including host-specificity shift, virulence, drug resistance, antigenic drift/escaped mutant are represented with triangles with different colours on each segment with the amino acid location and substitution change, and the number of samples that was observed (total n = 9). More detailed information regarding the mutation, which samples it occurred in, the reference sequence used, and any structural changes, can be found in Supplemental Table 4. HA mutation position nomenclature is based on the classical H3N2 strain.
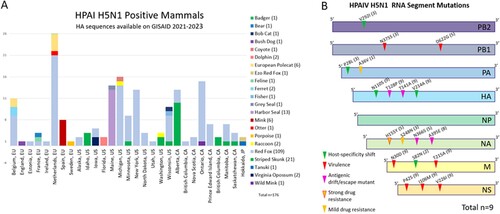
Information regarding previously reported mutations linked to host-specificity shift, virulence, drug resistance, and antigenic drift/escaped mutant in each of the eight genome segments is summarized in (B) and detailed in Supplemental Table 4. The PB2 segment recovered from three foxes presented the V292I mutation, which has been shown to result in host specificity shift [Citation36]. All five PB1 full length sequences obtained, showed the D622G mutation and three also presented the N375S mutation, which are both known to affect virulence [Citation37,Citation38]. The segment PA of three fox samples presented the mutation P28L, which has been linked to host specificity shift and mild drug resistance, and one had A36V, also associated with drug resistance [Citation39,Citation40]. The HA segment of the fox samples presented three important mutations found in all nine samples. Those include N101S, V214A, which have been associated with host-specificity shifts T141A, and T128P reported to result in antigenic drift / immune escape (classic H3N2 mutation nomenclature) [Citation41–45]. The NA segment of the fox HPAI viruses presented four different mutations; eight samples had A395E mutation, associated with antigenic drift/immune escape, while three samples also presented mutation S248N which affects antigenic drift / immune escape [Citation46]. Five fox HPAI sequences presented mutation A395E along with H155Y, which have been associated with strong drug resistance, and N366S, reported as antigenic drift /immune escape [Citation46–48]. All nine samples presented mutations N30D and T215A in the M1 protein, which affect virulence [Citation49]. Only two samples presented mutation S82N in the M2 protein, which affects host-specificity [Citation50,Citation51]. Finally, all nine fox HPAI samples presented mutation V2261, P42S, and I106M on NS1, which are known to contribute to virulence [Citation52–54].
All sequences recovered from the foxes here were determined to be HPAI H5N1 2.3.4.4b when the HA segment sequences were analysed and compared with various H5N1 clades from wild and domestic birds, 2015–2023. The fox derived HPAIV sequences appear to be closely related to viruses recovered from pheasants and domestic chickens in NY state (A).
Figure 7. (A) Phylogenetic tree shows that the samples tested in this study cluster with H5 clade 2.3.4.4b, using previously published clade annotations [Citation31,Citation55]. (B) Phylogenetic tree of worldwide mammalian HPAI H5N1 HA GISAID submissions (2021–2023). Sequence names presented from this study are highlighted in red and isolate identification simplified.
![Figure 7. (A) Phylogenetic tree shows that the samples tested in this study cluster with H5 clade 2.3.4.4b, using previously published clade annotations [Citation31,Citation55]. (B) Phylogenetic tree of worldwide mammalian HPAI H5N1 HA GISAID submissions (2021–2023). Sequence names presented from this study are highlighted in red and isolate identification simplified.](/cms/asset/6ff31558-8ede-4652-81e7-b260895f4b47/temi_a_2249554_f0007_oc.jpg)
The HA sequences from this study were analysed phylogenetically and compared to other sequences derived from mammals and collected worldwide, between 2021 and 2023 (B). Branches on the tree were collapsed and annotated with the geographic region where they were originally detected. The foxes in this study branch separately, with five samples clustering with other foxes detected in Michigan and Ontario, and California while the other four clustered with other foxes from Michigan and Iowa, a pair of opossums from Iowa, and sequences of HPAI detected in dolphins in Florida.
Conclusions
In this study, we present results on juvenile foxes in NYS that were found to be infected with HPAI subtype H5N1 clade 2.3.4.4b. Consistent with the neurological clinical signs observed in several of the affected foxes included here, high HPAIV loads were detected in the brain of the animals. This was confirmed with both IA RT-PCR and in-situ hybridization.
It is most probable that the fox kits acquired HPAI H5N1 2.3.4.4b through contact with or ingestion of infected wild birds. Experimental studies have previously shown that foxes become infected by feeding on infected bird carcasses [Citation6]. Phylogenetic analysis showed that the fox HPAI sequences were closely related to viruses recovered from pheasants and domestic chickens in NY state (A). Additionally, analysis of HPAI sequences derived from foxes from different states form different phylogenetic clusters, indicating multiple introductions in this animal species, likely through infected birds or waterfowl in their distinct geographic locations.
HPAI viral sequences have been recovered from red foxes from North America, Europe, and Asia (Ezo red fox in Japan) between 2021 and 2023. In addition to red foxes, HPAI has also been detected in many mammals, including seals, skunks, raccoons, opossums, bobcats, coyotes, fishers, and dolphins in North America. In Europe, HPAI H5N1 has been found in mink, ferrets, otter, feline, European polecats, porpoises, and badgers. It has also been found in a tanuki in Japan [Citation4,Citation8,Citation9,Citation24,Citation25,Citation55–57]. This highlights the relatively broad host range of the current HPAI H5NI strain and indicates that several carnivorous mammals or those that scavenger on dead animal carcasses should be considered at risk of infection.
While no evidence of direct or contact transmission has been reported in foxes, it is important to monitor infections in these species and continue to perform genomic sequencing of the virus in order to track mutations that may accumulate as the virus circulates in this highly susceptible mammalian host.
Supplemental Material
Download MS Word (47.7 KB)Acknowledgments
We want to acknowledge the staff at the AHDC, including Virology, Molecular Diagnostic, and Wildlife Health laboratories, as well as the USDA and other laboratories for submitting sequence data to GISAID. We also thank the NYS DEC and wildlife rehabilitators for collecting and submitting samples.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bosch FX, Orlich M, Klenk H-D, et al. The structure of the hemagglutinin, a determinant for the pathogenicity of influenza viruses. Virology. 1979;95:197–207. DOI:10.1016/0042-6822(79)90414-8
- Böttcher-Friebertshäuser E, Garten W, Matrosovich M, et al. The hemagglutinin: a determinant of pathogenicity. In: Compans RW, Oldstone MBA, editor. Influenza pathogenesis and control – volume I [Internet]. Cham: Springer International Publishing; 2014 [cited 2023 Jun 20]. p. 3–34. DOI:10.1007/82_2014_384
- Sagong M, Lee Y, Song S, et al. Emergence of clade 2.3.4.4b novel reassortant H5N1 high pathogenicity avian influenza virus in South Korea during late 2021. Transbounding Emerg Dis. 2022:tbed.14551.
- Bevins SN, Shriner SA, Cumbee JC, et al. Intercontinental movement of H5 2.3.4.4 highly pathogenic avian influenza A (H5N1) to the United States, 2021 [Internet]. Microbiology. 2022 [cited 2022 Jul 13]. Available from: http://biorxiv.org/lookup/doi/10.11012022.02.11.479922
- Richards B. Distribution of highly pathogenic avian influenza in North America, 2021/2022 [Internet]. [cited 2022 Jul 18]. Available from: https://www.usgs.gov/centers/nwhc/science/distribution-highly-pathogenic-avian-influenza-north-america-20212022
- Reperant LA, van Amerongen G, van de Bildt MWG, et al. Highly pathogenic avian influenza virus (H5N1) infection in red foxes fed infected bird carcasses. Emerg Infect Dis. 2008;14:1835–1841. DOI:10.3201/eid1412.080470
- Floyd T, Banyard AC, Lean FZX, et al. Encephalitis and death in wild mammals at a rehabilitation center after infection with highly pathogenic avian influenza A (H5N8) virus, United Kingdom. Emerg Infect Dis. 2021;27:2856–2863. DOI:10.3201/eid2711.211225
- Rijks JM, Hesselink H, Lollinga P, et al. Highly pathogenic avian influenza A (H5N1) virus in wild red foxes, The Netherlands, 2021. Emerg Infect Dis. 2021;27:2960–2962. DOI:10.3201/eid2711.211281
- Hiono T, Kobayashi D, Kobayashi A, et al. Virological, pathological, and glycovirological investigations of an Ezo red fox and a tanuki naturally infected with H5N1 high pathogenicity avian influenza viruses in Hokkaido, Japan. Virology. 2023;578:35–44. DOI:10.1016/j.virol.2022.11.008
- Kuiken T, Riteau B, Fouchier R, et al. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol. 2012;2:276–286. DOI:10.1016/j.coviro.2012.02.013
- Goodman LB, Anderson RR, Slater M, et al. High-throughput detection of respiratory pathogens in animal specimens by nanoscale PCR. JoVE. 2016: 54781.
- National Veterinary Services Laboratories NAHLN Document List [Internet]. USDA APHIS; 2022 [cited 2022 Sep 7]. Available from: https://www.aphis.usda.gov/animal_health/lab_info_services/downloads/ApprovedSOPList.pdf
- Shu B, Wu K-H, Emery S, et al. Design and performance of the CDC real-time reverse transcriptase PCR swine flu panel for detection of 2009 A (H1N1) pandemic influenza virus. J Clin Microbiol. 2011;49:2614–2619. DOI:10.1128/JCM.02636-10
- RNAscope 2.5 HD Detection Reagenet-Red User Manual [Internet]. Advanced Cell Diagnostics; [cited 2023 Mar 1]. Available from: https://acdbio.com/sites/default/files/322360-USM%20RNAscope%202.5%20HD%20RED%20Pt2_11052015.pdf
- Zhou B, Deng Y-M, Barnes JR, et al. Multiplex reverse transcription-PCR for simultaneous surveillance of influenza A and B viruses. McAdam AJ, editor. J Clin Microbiol. 2017;55:3492–3501. DOI:10.1128/JCM.00957-17
- Mitchell PK, Cronk BD, Voorhees IEH, et al. Method comparison of targeted influenza A virus typing and whole-genome sequencing from respiratory specimens of companion animals. J VET Diagn Invest. 2021;33:191–201. DOI:10.1177/1040638720933875
- Ip HS, Uhm S, Killian ML, et al. An evaluation of avian influenza virus whole-genome sequencing approaches using nanopore technology. Microorganisms. 2023;11:529. DOI:10.3390/microorganisms11020529
- Basecalling with Guppy — de.NBI Nanopore Training Course latest documentation [Internet]. [cited 2023 Apr 5]. Available from: https://denbi-nanopore-training-course.readthedocs.io/en/latest/basecalling/basecalling_1.html
- De Coster W, D’Hert S, Schultz DT, et al. Nanopack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. DOI:10.1093/bioinformatics/bty149
- Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. DOI:10.1186/s13059-019-1891-0
- Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature Communications. 2016;7:11257. DOI:10.1038/ncomms11257
- Suzuki H. minialign [Internet]. 2023 [cited 2023 Apr 5]. Available from: https://github.com/ocxtal/minialign
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. DOI:10.1093/bioinformatics/btr509
- Shi J, Zeng X, Cui P, et al. Alarming situation of emerging H5 and H7 avian influenza and effective control strategies. Emerg Microbes Infect; 12:2155072. DOI:10.1080/22221751.2022.2155072
- Cui P, Shi J, Wang C, et al. Global dissemination of H5N1 influenza viruses bearing the clade 2.3.4.4b HA gene and biologic analysis of the ones detected in China. Emerg Microbes Infect. 2022;11:1693–1704. DOI:10.1080/22221751.2022.2088407
- Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37:4572–4574. DOI:10.1093/bioinformatics/btab705
- Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008. DOI:10.1093/gigascience/giab008
- FluSurver [Internet]. Bioinformatics Institute; [cited 2023 Mar 17]. Available from: https://flusurver.bii.a-star.edu.sg/
- Argimón S, Abudahab K, Goater RJE, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genomics. 2016;2:e000093. DOI:10.1099/mgen.0.000093
- Geneious. Bioinformatics software for sequence data analysis [Internet]. Geneious. [cited 2023 Apr 9]. Available from: https://www.geneious.com/
- Zhang Y, Aevermann BD, Anderson TK, et al. Influenza research database: an integrated bioinformatics resource for influenza virus research. Nucleic Acids Res. 2017;45:D466–D474. DOI:10.1093/nar/gkw857
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. DOI:10.1093/molbev/mst010
- Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38:3022–3027. DOI:10.1093/molbev/msab120
- Releases rambaut/figtree [Internet]. GitHub. [cited 2023 Apr 5]. Available from: https://github.com/rambaut/figtree/releases
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526.
- Miotto O, Heiny A, Tan TW, et al. Identification of human-to-human transmissibility factors in PB2 proteins of influenza A by large-scale mutual information analysis. BMC Bioinformatics. 2008;9(Suppl 1):S18. DOI:10.1186/1471-2105-9-S1-S18
- Salomon R, Franks J, Govorkova EA, et al. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J Exp Med. 2006;203:689–697. DOI:10.1084/jem.20051938
- Feng X, Wang Z, Shi J, et al. Glycine at position 622 in PB1 contributes to the virulence of H5N1 avian influenza virus in mice. J Virol. 2016;90:1872–1879. DOI:10.1128/JVI.02387-15
- Khaliq Z, Leijon M, Belák S, et al. Identification of combinatorial host-specific signatures with a potential to affect host adaptation in influenza A H1N1 and H3N2 subtypes. BMC Genomics. 2016;17:529. DOI:10.1186/s12864-016-2919-4
- Mengual-Chuliá B, Alonso-Cordero A, Cano L, et al. Whole-genome analysis surveillance of influenza A virus resistance to polymerase complex inhibitors in eastern Spain from 2016 to 2019. Antimicrob Agents Chemother; 65:e02718-20.
- Naeem A, Elbakkouri K, Alfaiz A, et al. Antigenic drift of hemagglutinin and neuraminidase in seasonal H1N1 influenza viruses from Saudi Arabia in 2014 to 2015. J Med Virol. 2020;92:3016–3027. DOI:10.1002/jmv.25759
- Huang P, Sun L, Li J, et al. Potential cross-species transmission of highly pathogenic avian influenza H5 subtype (HPAI H5) viruses to humans calls for the development of H5-specific and universal influenza vaccines. Cell Discov. 2023;9:1–13.
- Watanabe Y, Ibrahim MS, Ellakany HF, et al. Acquisition of human-type receptor binding specificity by new H5N1 influenza virus sublineages during their emergence in birds in Egypt. PLoS Pathog. 2011;7:e1002068. DOI:10.1371/journal.ppat.1002068
- Nakajima S, Nakajima K, Nobusawa E, et al. Comparison of epitope structures of H3HAs through protein modeling of influenza A virus hemagglutinin: mechanism for selection of antigenic variants in the presence of a monoclonal antibody. Microbiol Immunol. 2007;51:1179–1187. DOI:10.1111/j.1348-0421.2007.tb04013.x
- Finkelstein DB, Mukatira S, Mehta PK, et al. Persistent host markers in pandemic and H5N1 influenza viruses. J Virol. 2007;81:10292–10299. DOI:10.1128/JVI.00921-07
- Saito T, Taylor G, Laver WG, et al. Antigenicity of the N8 influenza A virus neuraminidase: existence of an epitope at the subunit interface of the neuraminidase. J Virol. 1994;68:1790–1796. DOI:10.1128/jvi.68.3.1790-1796.1994
- Monto AS, McKimm-Breschkin JL, Macken C, et al. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob Agents Chemother. 2006;50:2395–2402. DOI:10.1128/AAC.01339-05
- Webster RG, Air GM, Metzger DW, et al. Antigenic structure and variation in an influenza virus N9 neuraminidase. J Virol. 1987;61:2910–2916. DOI:10.1128/jvi.61.9.2910-2916.1987
- Fan S, Deng G, Song J, et al. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology. 2009;384:28–32. DOI:10.1016/j.virol.2008.11.044
- Chen G-W, Shih S-R. Genomic signatures of influenza A pandemic (H1N1) 2009 virus. Emerg Infect Dis. 2009;15:1897–1903. DOI:10.3201/eid1512.090845
- Youk S-S, Lee D-H, Leyson CM, et al. Loss of fitness of Mexican H7N3 highly pathogenic avian influenza virus in mallards after circulating in chickens. J Virol. 2019;93. DOI:10.1128/jvi.00543-19.
- Jackson D, Hossain MJ, Hickman D, et al. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc Natl Acad Sci U S A. 2008;105:4381–4386. DOI:10.1073/pnas.0800482105
- Jiao P, Tian G, Li Y, et al. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82:1146–1154. DOI:10.1128/JVI.01698-07
- Ayllon J, Domingues P, Rajsbaum R, et al. A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J Virol. 2014;88:12146–12151. DOI:10.1128/JVI.01567-14
- Nagy A, Černíková L, Stará M. A new clade 2.3.4.4b H5N1 highly pathogenic avian influenza genotype detected in Europe in 2021. Arch Virol. 2022;167:1455–1459. DOI:10.1007/s00705-022-05442-6
- Re3data.Org. GISAID. 2012 [cited 2023 Apr 15]; Available from: https://www.re3data.org/repository/r3d100010126
- Bordes L, Vreman S, Heutink R, et al. Highly pathogenic avian influenza H5N1 virus infections in wild red foxes (Vulpes vulpes) show neurotropism and adaptive virus mutations [Internet]. Microbiology. 2022 [cited 2022 Jul 26]. Available from: http://biorxiv.org/lookup/doi/10.11012022.07.21.501071