
ABSTRACT
To characterize enteroviruses (EVs) circulating in farm animals in Central African Republic (CAR), we screened 192 stools of animals under 12 months belonging to family farms located in or near Bangui. To assess whether EV exchanges exist between these animals and humans, we also screened 195 stools of children who lived in contact with farm animals, as well as control stools of 358 children with no contact with farm animals. EVs were typed based on their capsid sequences.
In children, all EVs belonged to species A, B and C, with EV-Cs accounting for 60%. Some EV-Cs shared recent common ancestors with lineages of vaccine-derived poliovirus that emerged in the country in 2019–2020. In animals, we identified EV-Gs that belonged to 10 different types, including a previously unknown one that we named EV-G28, while no EV-E or EV-F were observed. The CAR EV-Gs were genetically closely related to specimens sampled in other continents and some of them harboured the torovirus-derived insertion already reported in some EV-Gs. The worldwide circulation of EV-Gs is likely due the massive international trade of live animals. Besides, two human EV-Cs (coxsackievirus A17 and coxsackievirus A24) were detected in pigs, suggesting that these viruses could cross the species barrier. Our work provides original data on the epidemiology and ecology of EVs circulating among herd animals in Africa.
Introduction
Enteroviruses (EVs) belong to the Picornaviridae family. Originally, EVs were classified into “serotypes”, each of which grouped together EV strains that were serologically indistinguishable from one another. Classification is now based on genetic relationships. Since the EV particles are naked, the capsid is the major determinant of phenotypic features, such as cell tropism and immunogenicity. Consequently, the current EV classification mainly relies on the capsid-encoding genomic region. Among the four proteins that compose the virus capsid (VP1 to VP4), VP1 bears the major neutralizing epitopes. For this reason, the VP1-encoding region is widely used to determine the virus types of EV field strains [Citation1,Citation2]. More than 300 virus types have been described and classified into 15 species: Enterovirus A to Enterovirus L and Rhinovirus A to Rhinovirus C [Citation3]. Besides EVs that circulate among humans, tens of EV types were detected in other mammals, including non-human primates (NHPs), livestock, camels and rodents [Citation4].
EV-Es, EV-Fs and EV-Gs infect livestock animals, such as swine, goats, sheep, cattle and lamas. Although the number of known specimens of EVs that infect livestock has increased during the last decade because of the introduction of next-generation sequencing techniques, these viruses remain under sampled, particularly in Africa were only few EV-Es, EV-Fs and EV-Gs have been reported [Citation5–7]. The respective host range of EV-Es, EV-Fs and EV-Gs is unknown. Infection of humans by EV-Es or EV-Fs have been suggested [Citation8,Citation9], but the specificity of the serological assays that were used has not been well-established. Nonetheless, one EV-E and one EV-F were detected in stools of NHPs, thus demonstrating that the transmission to primates can occur [Citation10]. The reverse transmission route is also possible, as illustrated by the swine vesicular disease virus (SVDV) and SVDV-2. Both are specific lineages of EV-Bs that circulate among humans, coxsackievirus B5 (CVB5) and CVB4, respectively. They established circulation among pigs in the 1950s or 1960s [Citation11,Citation12]. Therefore, EVs can cross the species barrier between primates and pigs, in both directions.
To characterize EV-Es, EV-Fs and EV-Gs circulating in farm animals in Central African Republic (CAR), stools of animals under 12 months (pigs, small ruminants and cattle) were molecularly screened and the EV genomes were sequenced by Illumina techniques. To determine whether animal EVs circulate in humans, stools of children living in close contacts with the farm animals were also screened, as were stools of control children with no contact with animals.
Material and methods
Collection of stool samples
Three groups were involved: i – domestic animals (calves, piglets and lambs) under 12 months of age in Bangui and surrounding areas, ii – children under 5 years of age living in close contact with/or nearby farm animals (“Contacts’), and iii – children under 5 years of age who do not live in close contact with animals (“Controls’). The latter were sampled amongst children coming for consultation at the emergency department of the Bangui Pediatric Complex. Stools were collected from September 2018 through December 2021. Animal stools were collected by anorectal swabs. After collection, all samples were stored in cooled boxes containing ice packs and transported to the laboratory where they were stored at – 80°C until processing.
Molecular screening
Anorectal swabs and stools were diluted in phosphate-buffered saline following the guidelines of the Polio Manual [Citation13]. Given the high number of samples, the suspensions were pooled before molecular screening.
RNAs from pooled suspensions were extracted using the ZYMO_RESEARCH® Kit. EV RNAs were detected using primers and probes (Supplementary Table 1) designed to target conserved nucleotide (nt) sequences within the 5’UTR of EV-Es, EV-Fs and EV-Gs. Because of the genetic variability in the 5’UTR, it was not possible to design a probe that matches with all EV-Es, EV-Fs and EV-Gs. Therefore, two probes were designed: one probe targeted 5’UTRs of EV-Es, – Fs and some EV-Gs (Assay A) found in goats and sheep while the second probe targeted most EV-Gs (Assay B).
The Taqman reactions were performed in a final volume of 20 µL by mixing 2 µL of extracted RNA with 10 µL of qScript XLT One-step RT-qPCR ToughMix (2×), 10 pmol of each primer, 5 pmol of the probe. The thermocycler profile was 50°C for 10 min, 95°C for 1 min followed by 40 cycles of PCR (95°C for 10 s, 60°C for 40s and 72°C for 30 s).
EV genome amplification and sequencing
The capsid-encoding region was amplified by using primers that target conserved genetic sequence among EV species in the 5’UTR (sense primers) and in the cis-replicating element (cre) located within the 2C-encoding region [Citation14,Citation15]. For this purpose, primers were designed based on EV-E, – F and – G genetic sequences retrieved from GenBank (Supplementary Table 1). Virus RNA was turned into cDNA by a RT step using the Maxima H Minus First Kit Strand cDNA Synthesis Kit (Thermo Scientific, ref K1652) with an Oligo(dT)18 primer: 4 µL of extracted RNA was mixed with 500 nM of each dNTP, 1 µL of primer (0.5 µg.µL−1) and 14 µL of nuclease-free water. The mixture was incubated at 65°C for 5 min and then on ice for 2 min; after adding 4 µL of RT buffer (5×) and 1 µL of Maxima H minus enzyme Mix, incubation was performed at 50°C for 30 min and 85°C for 5 min.
For amplifying the EV genomes prior to sequencing, the RT reaction was performed as described above by using random heptamers as primers. The cDNAs were used to amplify the 5’ half of the genomes and, for animal EVs, the 3’ half of the genomes. PCRs were carried out by using the Phusion High-Fidelity DNA polymerase (Thermo scientific).
To amplify the 5’ half of the genomes, a first PCR was performed using the cDNA as template with the primer pair EV-EFG-271-F/ EV-EFG-4441-R (Supplementary Table 1). Two µL of cDNA were mixed with 5 µL of Phusion HF Buffer (5×), 500 nM µL of each dNTP, 10 pmol of each primer, and 0.5 µL of enzyme. The thermocycler profile was: 98°C for 30 s followed by 35 cycles of amplification (98°C for 10 s, 50°C for 30 s and 72°C for 2 min) and a final extension step at 72°C for 10 min. If no band appeared on the agarose gel at the expected size, two nested PCRs were performed in parallel with the primer pairs EV-EF-381-F/ EV-EF-4357-R and EV-G-491-F/ EV-G-4294-R by using 2 µL of the product of the first PCR as template.
The 3’ half of the EV-G genomes was amplified by using the same protocol by using a sense primer that targets a conserved sequence within the 2C-encoding region and a reverse primer that targets the polyA tail (Supplementary Table 1).
The products of the PCR or nested PCR were purified on a NucleoFast®_96_PCR_Plate-DNA prior to sequencing by Illumina technology [Citation15]. Libraries were built using 0.1 ng of DNA with the Nextera XT DNA Library Preparation kit in a PCRmax Alpha Cycler 1 Thermal Cycler (Cole-Parmer). After purification on AMPure beads (Beckman), the libraries were controlled using the High Sensitivity D1000 assay (Agilent) on a TapeStation 4200 (Agilent). Sizing was achieved by electrophoresis on a PippinPrep System with the PippinPrep kit CDF1510 (Ozyme).
Sequence analysis
The Illumina raw sequences were analysed using CLC Genomics software (version 22). Contigs were built de novo and the contigs that correspond to EV genomic sequences were flagged through a BLAST analysis. Sequence alignments were performed with CLC Main Workbench (version 22) and phylogenetic trees were produced using MEGA X software [Citation16]. The phylogenetic trees were drawn by using the Minimum Evolution method. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. The minimum evolution tree was searched using the Close-Neighbour-Interchange (CNI) algorithm at a search level of 1. The Neighbour-joining algorithm was used to generate the initial tree. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test was calculated from 1,000 replicates.
Results
Origin of the samples
Animal samples were collected in farms located in four urban districts (4th, 6th, 7th and 8th arrondissements) of Bangui, the national capital and three other locations: Damara, a town located 75 km northeast of Bangui, Boali, a town located 55 km northwest of Bangui and Bouboui, which is between Bangui and Boali (Supplementary Figure 1). Overall, 192 animals were included in our study (): pigs represented 52% (99/192) of the animals, goats one third of the sampled animals while cattle and sheep represented less than 8% each (). The individual animal samples were pooled according to their respective animal species and location (Supplementary Table 2). The precise number of individual samples in each pool has not been recorded.
Figure 1. Characteristics of the three groups of subjects.
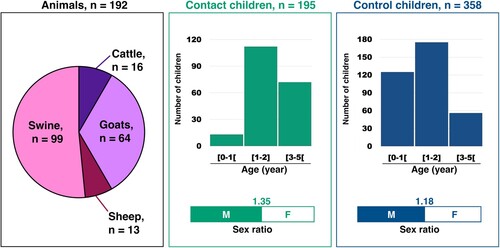
A total of 195 contact children and 358 control children under 5 years of age were included. The contact children lived in the places where the animals were sampled (Supplementary Table 3). As the control children were included in a paediatric hospital in Bangui, most of them came from Bangui and two areas that border Bangui, Bimbo and Bégoua (Supplementary Table 3 and Supplementary Figure 1). In both groups, around 50% of the children were 1–2 year-old (). The respective weight of the other age ranges differed between the two groups: children less than1 year old represented 35% of the control children while they were a small minority (6%) in the contact children (). Children over 3 years of age were more abundant in the contact group (36%) than in the control group (15%). The sex ratio was above 1 in both groups (). The stools of children were pooled according to their location and date of collection (Supplementary Table 3). The size of the pools ranged from 3 through 24 for contact children and from 7 to 12 for control children (Supplementary Figure 2). This difference is due to the different ways used to constitute the two groups: the contact children were sampled during field campaigns while the control children were recruited on a regular basis while they were consulting in the hospital.
Stools of pigs and goats contain mainly EV-Gs
After RNA extraction, EVs were detected through screening with a primer pair and two probes targeting the 5’UTR (). The primers and probes were designed to match with all known EV-E, – F and – G sequences but not to specifically target these EV species. The probe A gave a positive result for five pools from goats while the probe B was positive for one pool from goats and eight pools from swine. All pools from sheep and cattle were negative.
Figure 2. The upper scheme represents the enterovirus genome with the 5’ and 3’ untranslated regions (UTR) flanking the long open reading frame. The double arrows indicate the genomic regions that are amplified by the molecular assays used for screening (orange) and by the primers used for genome amplification before sequencing (blue and red). Below are indicated the number of pools that gave a positive result though molecular screening and genome amplification.
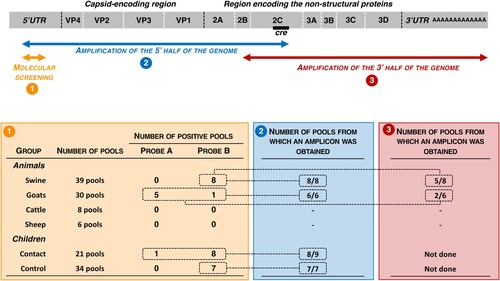
Since frequent recombination events shuffle EV genomes, molecular typing of EV field strains relies on the capsid-encoding genomic region, especially the VP1-encoding sequence [Citation17]. Two pairs of generic primers were used to perform RT–PCR and, when necessary, a subsequent nested-PCR. Both primer pairs target conserved sequences in both sides of the capsid-encoding region: the sense primers target the 5’UTR and the antisense primers targeted the cis-replicating element (cre) located within the 2C-encoding region (). The corresponding amplicons were ∼3,800 nt-long and encompassed the whole capsid and a large fraction of the 5’UTR and P2 genomic regions. All pools that gave a positive result through molecular screening were subjected to RT–PCR. Amplicons at the expected size were obtained for all of them ().
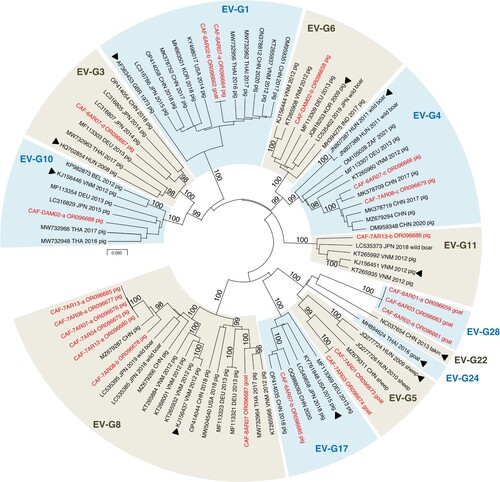
After Illumina sequencing and de novo assembly, the number of VP1 sequences per pool of animal stools ranged from 1 to 4. Overall, 23 contigs encompassing the VP1 sequences were identified, 21 of which belonged to the species EV-G and 2 to the species EV-C. The latter came from the same pool of swine samples and belonged to two virus types, CVA17 and CVA24. EV-Gs have been classified into 27 virus types (EV-G1 to EV-G27) based on VP1 relationships higher than 75% nt identity (N.J. Knowles, unpublished data). Among the 21 EV-G VP1 sequences of our dataset, 18 were assigned to 11 virus types already described (). Three contigs from goats did not cluster with any EV-G sequences available in public databases. Two encompassed the 5’UTR-2C genomic part (CAF-6AR02-a and CAF-6AR03, ∼4,140 nt-long) while the third one only covered the VP3-2C region (CAF-6AR01-a, 2,169 nt). They differed by only few nt differences from one another. In the VP1-encoding region, they displayed less than 74% nt identity with other known EV-Gs, which is below the threshold used to classified two strains in the same virus type. The closest match was the strain LLT03 [Citation18], which is the only known representative of the EV-G22 virus type (Supplementary Figure 3). In the VP1-encoding region, CAF-6AR02-a, CAF-6AR03 and CAF-6AR01-a displayed with LLT03 an identity lower than 72% at nt level and lower than 83% at amino-acid level. Therefore, they were assigned to a new type, called “EV-G28”.
Figure 3. Phylogenetic relationships between CAR EV-Gs and highest similarity sequences identified in public database, based on the VP1-encoding region. CAR specimens are in red. The other sequences are named by using their respective GenBank accession number, country (ISO 3166-1 alpha-3 codes), year of sampling and host, when known. Triangles indicate the prototype strain of each virus type. Bootstrap values are indicated if higher than 95%. For a better readability, only relevant virus types are shown. A tree featuring all known EV-G virus types is provided as Supplementary Figure 3.
EV-Gs found in CAR are genetically close to EV-Gs sampled in other continents
To study the genetic relationships of the EVs detected in this study with previously reported EVs, two genomic regions were considered independently: the 5’UTR and the 2A-cre genomic region, which are located upstream and downstream the capsid-encoding region, respectively. The Basic Local Alignment Search Tool (BLAST) was used to find genetic sequences with high similarity and draw phylogenetic trees. In both regions, sequences did not cluster by virus types, indicating that intertypic recombination takes place between EV-Gs in both sides of the capsid-encoding region ().
Figure 4. Phylogenetic relationships between CAR EV-Gs and highest similarity sequences identified in public database, based on the 5’UTR and 2A-cre region. CAR specimens are in red. The other sequences are named by using their respective GenBank accession number, country (ISO 3166-1 alpha-3 codes), year of sampling and host, when known. Triangles indicate the prototype strain of each virus type. Some groups have been collapsed for a better legibility. Bootstrap values are indicated if higher than 90%.

It is known that EV-G 5’UTR sequences fall into two distinct groups [Citation4]. One group (group A in ) only contains sequences of EV-Gs and gathers all known EV-Gs from swine; the second one (group B in ) is composed of EV-G sequences that are phylogenetically and structurally close to the 5’UTRs of EV-Es and EV-Fs. All the CAR EV-G 5’UTR sequences from pigs belonged to the first group, which also contained two sequences of CAR EVs found in goats (EV-G1 CAF-6AR02-b and EV-G8 CAF-8AR707). The four other CAR EV-G 5’UTR sequences from goats belonged to the second group and were closely related to that of an EV-G20 sampled in Austria in 2019 [Citation19].
The clustering observed in the 2A-cre region was congruent with that of the 5’UTR sequences: the viruses whose 5’UTR sequences belonged to group A clustered with EV-Gs of different types sampled in pigs and wild boars while viruses whose 5’UTR sequences belonged to group B fell into a separate branch with EV-Gs sampled in goats, sheep and takins ().
Interestingly, the 5’UTR and 2A-cre sequences of EV-Gs identified here were closely related to those of viruses sampled in different continents (). CAR EV-Gs were closely related to EV-Gs previously sampled in Asia and Europe (). Thus, the EV-Gs that circulate in farm animals in CAR do not form specific genetic lineages, which suggests a global circulation of EV-Gs.
Some EV-Gs found in CAR display an insertion in their genome
Since 2017, EV-G genomes that carry an insertion at the 2C-3A junction have been repeatedly detected in different countries [Citation4]. This insertion is phylogenetically related to papain-like cysteine protease (PL-CP) sequences of swine toroviruses (family Tobaniviridae, genus Torovirus) and was probably acquired through recombination. To determine whether this insertion was present in the genome of some CAR EV-Gs, we amplified the second half of the virus genome () by using a primer pair that targets a conserved region within the 2B-encoding genomic region (sense primer) and the poly – A tail (antisense primer). Eight contigs were thus obtained from 7 pools of animal stools (5 from swine and 2 from goats), while no amplicons were obtained for the 11 other pools (). Seven animal pools generated one single contig that could be assembled to 5’ half genomic sequences found in the corresponding pools, thus forming contigs that spanned virtually the full-length genome (). Five of them (EV-G1 CAF-6AR07-a, EV-G17 CAF-6AR07-b, EV-G8 CAF-7AR04, EV-G8 CAF-7AR12-a and EV-G8 CAF-7AR13-a), all from pigs, contained an insertion whereas two from goats (EV-G5 CAF-7AR01, EV-G5 CAF-7AR03) did not. One pool (from pig) contained three distinct contigs that spanned the 2C-3A region; only one (CAF-6AR08-b) contained an insertion, which was similar to the one found in EV-G1 CAF-6AR07-a.
Table 1. Presence or absence of a PL-CP insertion in EV-G non-structural genomic region.
The 6 insertions were aligned with PL-CP insertions previously reported. All insertions found in this study were relatively close to one another and were related to PL-CP sequences reported in EV-Gs samples in China between 2019 and 2021 (). These results confirm that CAR pigs are infected with EV-Gs that circulate at global scale.
Figure 5. Dendrogram based on the PL-CP insertion found in some EV-G specimens. CAR specimens are in red. The other sequences are named by using their respective GenBank accession number, country (ISO 3166–1 alpha-3 codes), year of sampling and host, when known. A sequence of a torovirus PL-CP was used for rooting the tree. Bootstrap values are indicated if higher than 95%.
Stools of children do not contain animal EVs
Pools of human samples were screened with the same molecular assays as those used to screen the farm animal samples. The highest positive rate was observed for the pools of the contact children, of which 9 out of 21 (42.9%) were positive with at least one test (Supplementary Table 3). One pool was positive with the probe A, eight with the probe B (). This rate was around 20% for the children of the control groups, of which 7 out of 34 were positive with the probe B. The result of the molecular screening was not linked to the number of individual samples contained in the pools (Supplementary Figure 2).
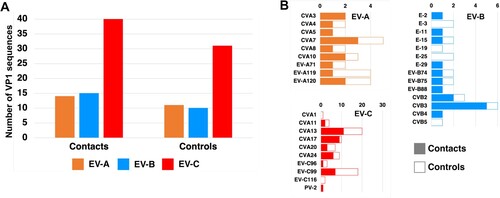
After amplification and Illumina sequencing, the number of VP1 sequences per human pool ranged from 5 to 16. Overall, 126 contigs encompassing the VP1-encoding genomic region were generated from human pools. They belonged to the species EV-A, EV-B and EV-C ((A)). No animal EV was detected in the human samples. The repartition was similar in both groups (contacts and controls): the members of species EV-C accounted for 58% of the viruses, while EV-As and EV-Bs represented 21% each. Thirty-three virus types were found in these samples ((B)). CVA13 and EV-C99, two members of the species EV-Cs, were by far the most abundant viruses since they represented together around a third of the viruses that were detected in children. One PV-2 was detected, which derived from the type 2 vaccine strain. It displayed 6 nt differences with this strain in the VP1-encoding region, which is the threshold used to differentiate vaccine-derived PVs (VDPVs) from Sabin-like strains. In summary, no differences were observed between the virus types found in the two groups of children. These results suggest that EVs that circulate in farm animal do not transmit to humans, even those living on farm premises.
Figure 6. Distribution of the EVs found in child stools into species (A) and virus types (B).
Some EV-Cs found in human samples are genetically related to vaccine-derived polioviruses
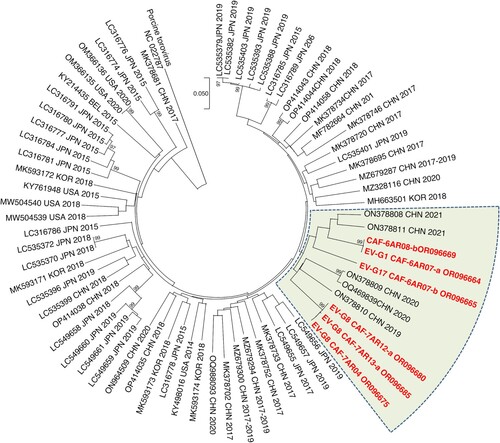
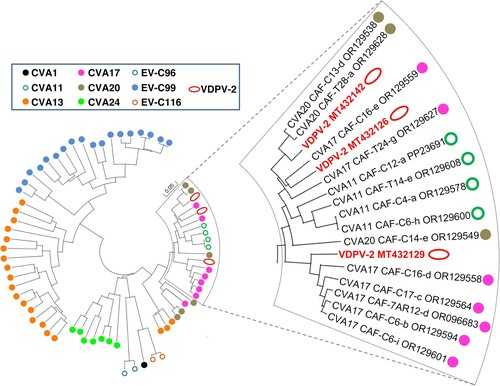
The BLAST analysis revealed that some identified EV-Cs feature a high nt identity with vaccine-derived polioviruses (VDPVs) that emerged in CAR in 2019–2020 [Citation20]. Most of these VDPVs had recombinant genomes made of genetic sequences coming from the PV-2 vaccine strain (at least the 5’UTR and the capsid-encoding region) and sequences from non-polio EVs (NPEVs) [Citation20]. No close relatives to the NPEV sequences harboured by these VDPVs had previously been found in public databases. Two CVA20s sampled in children during this study featured a nt identity higher than 94% in the 2A-cre region with a group of VDPVs of which CAF-19-RS7-MMP-PGP-005-B5 is a representative (), which suggests that these CVA20s and the VDPVs of this group shared a recent common ancestor through recombination. Similarly, one CVA17 (CAF-C16-e) displayed a nt identity higher than 91% in this region with CAF-19-RS4-KEM-103-C1 whereas another set of CVA17 was very close to CAF-19-RS4-KEM-170 (nt identity higher than 96%). Thus, some EV-Cs were very close to those that exchanged genetic material with the poliovirus vaccine strain and gave rise to the VDPV-2 lineages that emerged in the country in 2019.
Figure 7. Phylogenetic relationships between CAR EV-Cs and some vaccine-derived poliovirus isolates, based on the 2A-cre genomic region. Specimens are colour-coded according to their respective virus type. The branch containing the vaccine-derived poliovirus isolates is magnified in the right panel.
Discussion
This study aimed to identify EV-Es, EV-Fs and EV-Gs that circulate among farm animals in CAR and to determine whether these viruses infect children who live in close contact with these animals. The absence of detection of EV-Es and EF-Fs could be attributed to the lack of sensitivity of the assay we designed to target EV-E and EV-F 5’UTR sequences (probe A). This is however unlikely since this probe detected EV-Gs whose 5’UTR is close to that of EV-Es and EV-Fs. This result could also be due to the small number of goats and sheep included in our study. Nonetheless, in another study, the screening of 164 rectal swabs from goats and sheep living in Gabon did not reveal EV-Es or EV-Fs either [Citation7]. It is difficult to interpret these data since the prevalence of EV-Es and EV-Fs in healthy farm animals is unknown in Africa. Few serological studies revealed high seroprevalence among different mammal species [Citation8,Citation9,Citation21], but none of these studies were conducted in Africa. Furthermore, the specificity of the serological assays has never been evaluated. Previous genomic studies reported relatively high detection rate: 59% of the screened animals were infected by a member of the species EV-E in Colombia [Citation22], and 39.5% of the cattle and 47% of the goats were found positive in Thailand [Citation23]. Therefore, new studies involving larger numbers of sheep and goats must be undertaken to determine whether EV-Es and EV-Fs circulate among farm animals in Central Africa.
In the past, two EV-Bs that infect humans, namely CVB4 and CVB5, were able to cross the species barrier to establish sustained transmission among swine [Citation4,Citation12,Citation24]. To our knowledge, capability of EV-Cs to infect swine has never been demonstrated. It is known that CVA17 and CVA24 (the two virus types that have been detected in swine samples in our work) can use the human intercellular adhesion molecule-1 (ICAM-1) as cellular receptor [Citation25,Citation26]. Swine and human ICAM-1 only display 53% identity at amino-acid level with one another [Citation27] and the residues that have been identified in human ICAM-1 to interact with CVA21 particles are poorly conserved in swine ICAM-1 [Citation28]. However, it has been shown that some CVAs of the species EV-C were able to use alternate receptors [Citation29]. It is therefore possible that infection of pigs by these viruses is mediated by receptors that remain to be identified. Alternatively, it could be passive carriage of human EVs by pigs after drinking contaminated water or even eating human stools, since pigs are coprophagous. It is noteworthy that one EV-B80 and one EV-C99 were previously reported in animal stools collected in Gabon, from a goat and a dog respectively [Citation7]. It is therefore possible that some human EVs infect other hosts, even though the low number of human EVs detected in animal pools suggest that such infections are uncommon.
At least two sub-groups exist within the species EV-G, one made of viruses that circulate among swine and another made of viruses that circulate among goats, sheep and takins. Both sub-groups feature capsid-encoding sequences that remain genetically close together while their non-capsid regions segregate into two separate clusters, depending on their respective hosts. Similar observations were previously made within species EV-A and EV-B [Citation4], of which some members that circulate among NHPs are closer to simian EVs of other EV species than to human EVAs or EV-Bs in the non-structural region of their genomes [Citation30–32]. These data further demonstrate that the forces that drive the EV evolution are not evenly applied along their genome. As previously suggested [Citation33], EV evolution is a modular process in which the 5’UTR and the non-structural regions evolve somewhat independently from the capsid-encoding region. Previous observations made on EV-As and EV-Bs and the differential phylogenetic clustering we observed for EV-Gs depending on their respective host strongly suggest that the 5’UTR and the non-structural regions are major determinants of the adaptation to host. Thus, crossing the barrier species by EVs could involve recombination events that generate new genomes by associating the capsid-encoding region of an EV of a given species to non-capsid regions of EVs belonging to another species well-adapted to the new host. In this light, the detection of one EV-G1 (CAF-6AR02-b) and one EV-G8 (CAF-8AR07) in goat stools is surprising. To our knowledge, these two EV-G types have never been detected in animals other than pigs or wild boars. These two specimens feature all the genetic treats of swine EV-Gs, including the absence of the 5’UTR generally found in EV-Gs that infect goats, sheep and takins. Therefore, this observation must be considered with caution until other studies confirm the presence of EV-G1 and EV-G8 in goats. For instance, cross-contamination of the goat samples could have occurred when the stools were pooled. However, sporadic infection of goats by EVs that circulate among pigs could take place though contaminated drinking water [Citation23].
The close genetic relationships of EV-Gs found in CAR with EV-Gs previously sampled in various regions of the world suggest that EV-Gs that infect livestock form a single worldwide ecosystem. This can be explained by the high number of live pigs that are exported every year. According to the Food and Agriculture Organization of the United Nations trade dataset (available at https://www.fao.org/faostat/en/#data/TM/visualize), Middle African countries (Angola, Cameroon, CAR, Democratic Republic of the Congo, Gabon and São Tomé and Príncipe) imported live pigs from different European countries and from the United States of America from 2017 through 2022. The trade of livestock is a major risk of transboundary spread of diseases, as already demonstrated by the global dissemination of the African Swine fever from its original cradle in Africa [Citation34].
No animal EVs were detected in human samples. As the limit of detection of the assays we used to screen the stools is unknown, it is not possible to rule out the presence of animal EVs in human stools that could have remained undetected because the samples were pooled. Nonetheless, this result suggests that human infections by EV-Gs are unusual, even in children that live in close contacts with farm animals. While the primers used for the molecular screening were designed based on EV-E, EV-F and EV-G genetic sequences, members of species EV-A, -B and -C were detected in human pools, which is not surprising given the high level of genetic conservation among the 5’UTRs of different EV species. Interestingly, no EV-Ds were detected, although EV-D94 and EV-D111 were previously found in human samples collected in sub-Saharan Africa [Citation30,Citation35–37]. This result suggest that these two virus types remain relatively uncommon in humans, even in this region of the world where they were originally sampled [Citation37,Citation38].
The predominance of EV-Cs in human samples agrees with previous studies reporting a relatively high abundance of EV-Cs in children in sub-Saharan Africa [Citation39–42]. This seems to be a specific treat of the EV ecosystem in this part of the world since EV-Cs are minority in North America, Europe and Asia. One “young” VDPV-2 (i.e. a VDPV with a low number of nt differences compared to the vaccine strain) was also detected. This is not surprising since the collections of human samples took place when multiple lineages of VDPV-2 were actively circulating in the country [Citation20]. Numerous vaccination campaigns were then conducted with the monovalent oral polio vaccine 2, thus explaining how a child was infected by a VDPV-2 still closely related to the vaccine strain. Furthermore, we identified some EV-Cs that shared recent common ancestors with the VDPV-2 lineages that emerged in CAR few months before our sample collections [Citation20]. Launched in 2021, the novel oral polio vaccine (nOPV) was designed to reduce the risk of reversion. Nonetheless, recombination between nOPV and circulating EV-Cs is still possible, as already observed in some countries, including CAR [Citation43]. Therefore, the active circulation of EV-Cs that can exchange genetic material with vaccine PV strains constitutes a major risk factor of reversion and should be monitored.
In conclusion, our study conducted in CAR revealed that EV-Gs that are genetically linked to strains sampled in other continents circulate in pigs and goats in this country. Animal EVs were not detected in children living in close contact with the infected animals, which suggest that transmission of EV-Gs from farm animals to humans is uncommon, even in countries where sanitation is poorly developed. However, the detection of two EV-Cs in swine samples and of one EV-G1 and one EV-G8 in goats suggest that cross-species transmission can sporadically occur from humans to farm animals and from swine to goats.
Ethics
This study was conducted in compliance with the Central African regulations, the Declaration of Helsinki, the provisions relating to biomedical research of the Council for International Organizations of Medical Sciences, and the research protocol. We have obtained the ethical agreement of the Ethics and Scientific Committee of the University of Bangui and the Institut Pasteur de Bangui.
Supplemental Material
Download MS Word (445.3 KB)Supplemental Material
Download MS Word (386.8 KB)Supplemental Material
Download MS Word (2.8 MB)Supplemental Material
Download MS Word (15.5 KB)Supplemental Material
Download MS Word (15.3 KB)Supplemental Material
Download MS Word (16.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Oberste MS, Maher K, Flemister MR, et al. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J Clin Microbiol. 2000;38:1170–1174. doi:10.1128/JCM.38.3.1170-1174.2000
- Oberste MS, Maher K, Kilpatrick DR, et al. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999;73:1941–1948. doi:10.1128/JVI.73.3.1941-1948.1999
- Simmonds P, Gorbalenya AE, Harvala H, et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch Virol. 2020;165:793–797. doi:10.1007/s00705-019-04520-6
- Sadeuh-Mba S, Doté J, Joffret M-L, et al. Animal enteroviruses: a glimpse of a wide evolutionary landscape. Virologie (Montrouge). 2023;27:22–34. doi:10.1684/vir.2023.1000
- Faleye TOC, Adewumi OM, Olayinka OA, et al. Draft genome sequence of a bovine enterovirus isolate recovered from sewage in Nigeria. Microbiol Resour Announc. 2018;7:e01466–18.
- Sobhy NM, Mor SK, Mohammed MEM, et al. Isolation and molecular characterization of bovine enteroviruses in Egypt. Vet J. 2015;206:317–321. doi:10.1016/j.tvjl.2015.10.011
- Bohou Kombila L, N’dilimabaka N, Garcia D, et al. Molecular identification of enteric viruses in domestic animals in northeastern Gabon, Central Africa. Animals (Basel). 2023;13:2512. doi:10.3390/ani13152512
- Gür S, Gürçay M, Seyrek A. A study regarding bovine enterovirus type 1 infection in domestic animals and humans: an evaluation from the zoonotic aspect. J Vet Med Sci. 2019;81:1824–1828. doi:10.1292/jvms.18-0704
- Gür S, Yapkiç O, Yilmaz A. Serological survey of bovine enterovirus type 1 in different mammalian species in Turkey. Zoonoses Public Health. 2008;55:106–111. doi:10.1111/j.1863-2378.2007.01095.x
- Oberste MS, Feeroz MM, Maher K, et al. Characterizing the picornavirus landscape among synanthropic nonhuman primates in Bangladesh, 2007 to 2008. J Virol. 2013;87:558–571. doi:10.1128/JVI.00837-12
- Knowles NJ, McCauley JW. Coxsackievirus B5 and the relationship to swine vesicular disease virus. Curr Top Microbiol Immunol. 1997;223:153–167.
- Lomakina NF, Shustova Y, Strizhakova E, et al. Epizootic of vesicular disease in pigs caused by coxsackievirus B4 in the Soviet Union in 1975. J Gen Virol. 2016;97:49–52. doi:10.1099/jgv.0.000318
- Anonymous. 6.2 Specimen préparation. Polio laboratory manual [Internet]. 4th edition Geneva: World Health Organization; 2004; p. 82–83. Available from: https://apps.who.int/iris/bitstream/handle/10665/68762/WHO_IVB_04.10.pdf?sequence=1&isAllowed=y.
- Bessaud M, Jegouic S, Joffret M-L, et al. Characterization of the genome of human enteroviruses: design of generic primers for amplification and sequencing of different regions of the viral genome. J Virol Methods. 2008;149:277–284. doi:10.1016/j.jviromet.2008.01.027
- Joffret M-L, Polston PM, Razafindratsimandresy R, et al. Whole genome sequencing of enteroviruses species A to D by high-throughput sequencing: application for viral mixtures. Front Microbiol. 2018;9:2339. doi:10.3389/fmicb.2018.02339
- Kumar S, Stecher G, Li M, et al. Mega X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi:10.1093/molbev/msy096
- Oberste MS, Maher K, Kilpatrick DR, et al. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999;73:1941–1948. doi:10.1128/JVI.73.3.1941-1948.1999
- Guan T-P, Teng JLL, Yeong K-Y, et al. Metagenomic analysis of Sichuan takin fecal sample viromes reveals novel enterovirus and astrovirus. Virology. 2018;521:77–91. doi:10.1016/j.virol.2018.05.027
- Weissenböck H, Ebinger A, Gager AM, et al. A novel enterovirus in lambs with poliomyelitis and brain stem encephalitis. Transbound Emerg Dis. 2022;69:227–234. doi:10.1111/tbed.14412
- Joffret M-L, Doté JW, Gumede N, et al. Vaccine-derived polioviruses, Central African Republic, 2019. Emerg Infect Dis. 2021;27:620–623. doi:10.3201/eid2702.203173
- Erol N, Gür S, KoÇ BT, et al. A serological investigation of Bovine enterovirus-1, Bovine herpesvirus-1, Bovine viral diarrhea virus, and Parainfluenza-3 infections in camelsin Western Turkey. Vet Ital. 2020;56:257–262.
- Medina JE, Castañeda S, Páez-Triana L, et al. High prevalence of Enterovirus E, Bovine Kobuvirus, and Astrovirus revealed by viral metagenomics in fecal samples from cattle in Central Colombia. Infect Genet Evol. 2024;117:105543. doi:10.1016/j.meegid.2023.105543
- Income N, Kosoltanapiwat N, Taksinoros S, et al. Molecular identification of enteroviruses from cattle and goat feces and environment in Thailand. Appl Environ Microbiol. 2019;85:e02420–18. doi:10.1128/AEM.02420-18
- Zhang G, Wilsden G, Knowles NJ, et al. Complete nucleotide sequence of a coxsackie B5 virus and its relationship to swine vesicular disease virus. J Gen Virol. 1993;74(Pt 5):845–853. doi:10.1099/0022-1317-74-5-845
- Baggen J, Hurdiss DL, Zocher G, et al. Role of enhanced receptor engagement in the evolution of a pandemic acute hemorrhagic conjunctivitis virus. Proc Natl Acad Sci U S A. 2018;115:397–402. doi:10.1073/pnas.1713284115
- Jegouic S, Joffret M-L, Blanchard C, et al. Recombination between polioviruses and co-circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 2009;5:e1000412. doi:10.1371/journal.ppat.1000412
- Stocker CJ, Sugars KL, Yarwood H, et al. Cloning of porcine intercellular adhesion molecule-1 and characterization of its induction on endothelial cells by cytokines1. Transplantation. 2000;70:579–586. doi:10.1097/00007890-200008270-00008
- Xiao C, Bator CM, Bowman VD, et al. Interaction of coxsackievirus A21 with its cellular receptor, ICAM-1. J Virol. 2001;75:2444–2451. doi:10.1128/JVI.75.5.2444-2451.2001
- Newcombe NG, Andersson P, Johansson ES, et al. Cellular receptor interactions of C-cluster human group A coxsackieviruses. J Gen Virol. 2003;84:3041–3050. doi:10.1099/vir.0.19329-0
- Harvala H, Sharp CP, Ngole EM, et al. Detection and genetic characterization of enteroviruses circulating among wild populations of chimpanzees in Cameroon: relationship with human and simian enteroviruses. J Virol. 2011;85:4480–4486. doi:10.1128/JVI.02285-10
- Mombo IM, Lukashev AN, Bleicker T, et al. African non-human primates host diverse enteroviruses. PLoS One. 2017;12:e0169067. doi:10.1371/journal.pone.0169067
- Sadeuh-Mba SA, Bessaud M, Joffret M-L, et al. Characterization of enteroviruses from non-human primates in Cameroon revealed virus types widespread in humans along with candidate new types and species. PLoS Negl Trop Dis. 2014;8:e3052. doi:10.1371/journal.pntd.0003052
- Muslin C, Mac Kain A, Bessaud M, et al. Recombination in enteroviruses, a multi-step modular evolutionary process. Viruses. 2019;11:859. doi:10.3390/v11090859
- Li Z, Chen W, Qiu Z, et al. African swine fever virus: a review. Life (Basel). 2022;12:1255.
- Chern S-WW, Gumede N, Castro CJ, et al. Whole-genome sequences of enteroviruses D94 and D111 isolated from stool specimens in Angola. Microbiol Resour Announc. 2021;10:e00728–21.
- Majumdar M, Klapsa D, Wilton T, et al. High diversity of human non-polio enterovirus serotypes identified in contaminated water in Nigeria. Viruses. 2021;13:249. doi:10.3390/v13020249
- Sadeuh-Mba SA, Joffret M-L, Mazitchi A, et al. Genetic and phenotypic characterization of recently discovered enterovirus D type 111. PLoS Negl Trop Dis. 2019;13:e0007797.
- Smura TP, Junttila N, Blomqvist S, et al. Enterovirus 94, a proposed new serotype in human enterovirus species D. J Gen Virol. 2007;88:849–858. doi:10.1099/vir.0.82510-0
- Adeniji JA, Oragwa AO, George UE, et al. Preponderance of enterovirus C in RD-L20B-cell-culture-negative stool samples from children diagnosed with acute flaccid paralysis in Nigeria. Arch Virol. 2017;162:3089–3101. doi:10.1007/s00705-017-3466-2
- Bessaud M, Pillet S, Ibrahim W, et al. Molecular characterization of human enteroviruses in the Central African Republic: uncovering wide diversity and identification of a new human enterovirus A71 genogroup. J Clin Microbiol. 2012;50:1650–1658. doi:10.1128/JCM.06657-11
- Osundare FA, Opaleye OO, Akindele AA, et al. Detection and characterization of human enteroviruses, human cosaviruses, and a new human parechovirus type in healthy individuals in Osun State, Nigeria, 2016/2017. Viruses. 2019;11:1037. doi:10.3390/v11111037
- Sadeuh-Mba SA, Bessaud M, Massenet D, et al. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J Clin Microbiol. 2013;51:759–770. doi:10.1128/JCM.02119-12
- Davlantes E, Jorba J, Henderson E, et al. Notes from the field: circulating vaccine-derived poliovirus type 2 emergences linked to novel oral poliovirus vaccine Type 2 use – six African countries, 2021–2023. MMWR Morb Mortal Wkly Rep. 2023;72:1041–1042. doi:10.15585/mmwr.mm7238a4