
ABSTRACT
SARS-CoV-2 has been evolving into a large number of variants, including the highly pathogenic Delta variant, and the currently prevalent Omicron subvariants with extensive evasion capability, which raises an urgent need to develop new broad-spectrum neutralizing antibodies. Herein, we engineer two IgG-(scFv)2 form bispecific antibodies with overlapping epitopes (bsAb1) or non-overlapping epitopes (bsAb2). Both bsAbs are significantly superior to the parental monoclonal antibodies in terms of their antigen-binding and virus-neutralizing activities against all tested circulating SARS-CoV-2 variants including currently dominant JN.1. The bsAb1 can efficiently neutralize all variants insensitive to parental monoclonal antibodies or the cocktail with IC50 lower than 20 ng/mL, even slightly better than bsAb2. Furthermore, the cryo-EM structures of bsAb1 in complex with the Omicron spike protein revealed that bsAb1 with overlapping epitopes effectively locked the S protein, which accounts for its conserved neutralization against Omicron variants. The bispecific antibody strategy engineered from overlapping epitopes provides a novel solution for dealing with viral immune evasion.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the pathogen of COVID-19 [Citation1], utilizing its spike (S) protein to bind the human angiotensin-converting enzyme 2 (ACE2) receptor and invade cells [Citation2,Citation3]. During transmission, it rapidly evolves numerous variants (Supplementary Fig. 1a), some of which are significantly more transmissible or pathogenic, or show significant immune escape, resulting in a diminished efficacy of existing therapeutic and prophylactic measures, such as the previously circulating variants of concern (VOCs) Alpha [Citation4], Beta [Citation5], Gamma [Citation6], and Delta [Citation7]. At the end of 2021, Omicron emerged and spread rapidly with more than 30 mutations in its S protein, 15 of which occurred in the receptor-binding domain (RBD) [Citation8,Citation9]. Soon after the appearance of BA.1, many subvariants have evolved and given rise to multiple global infection waves [Citation10–13].
Several studies reported that Omicron and its subvariants dramatically escape the neutralization of sera from convalescent or vaccinated individuals [Citation14–20]. In addition, the vast majority of the antibodies, including most previously approved monoclonal antibodies (mAbs) or cocktail therapies, were found to have extensively reduced or even completely lost their neutralizing activity against these viral strains [Citation9,Citation14,Citation15,Citation21–24]. For instance, the combination of casirvimab (REGN10933) with imdevimab (REGN10987) and bamlanivimab (LY-CoV555) plus etesevimab (LY-CoV016/CB6) have been shown to lose efficacy against BA.1 [Citation21,Citation25,Citation26]. Compared to the D614G strain, sotrovimab (S309) was approximately 12-fold and 10-fold less potent in neutralizing BA.2 and BA.4, respectively [Citation26]. Moreover, evusheld, the cocktail of tixagevimab (COV2-2196/AZD8895) and cilgavimad (COV2-2130/AZD1061) was revoked emergency use authorization (EUA) by the US Food and Drug Administration (FDA) due to the inability to neutralize XBB and XBB.1.5 [Citation17,Citation23,Citation27], and another approved antibody bebtelovimab (LY-CoV1404) was evaded by the XBB.1, XBB.1.5, and BQ.1.1 [Citation23,Citation27]. JN.1, with just one additional mutation (L455S) compared to its predecessor BA.2.86, was reported having ability to evade class 1 antibodies [Citation12]. The remarkable immune escape and the further evolution of the virus underscore the importance of developing new next-generation and potent cross-protective therapeutics and vaccines.
As a new therapeutic strategy, multi-modality bispecific antibodies (bsAbs) against SARS-CoV-2 have been developed recently, like the IgG – VHH format of SYZJ001 [Citation28], IgG-like FD01 [Citation29], and DVD-Ig format of CV1206_521_GS [Citation30]. Among them, IgG-(scFv)2 format shows excellent capacity due to its four binding moieties and its low susceptibility to antibody-dependent enhancement (ADE) [Citation31,Citation32]. Besides, bsAbs with the Fc region like the IgG-(scFv)2 format have a prolonged half-life and stability in vivo and retain Fc-mediated effector functions, such as antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and antibody-dependent cellular phagocytosis (ADCP) [Citation33,Citation34]. In drug development, bispecific antibodies are a more cost-effective strategy than antibody cocktails which require the production of two separate antibodies. Moreover, it has been found that bispecific antibodies are more capable of inhibiting the generation of drug-resistant mutations than monoclonal or cocktail antibodies, which has important implications for clinical therapy [Citation32].
In general, there is a tendency to select antibodies with different binding epitopes as parent antibodies, while bispecific antibodies designed from overlapping epitopes have been less studied. In this study, we used three previously discovered neutralizing antibodies to engineer two IgG-(scFv)2 form bispecific antibodies bsAb1 and bsAb2. BsAb1 is constructed from H4B6 and H4D12 that recognize overlapping epitopes, whereas bsAb2 is composed of the noncompeting H4D12 and ZW2G10. We demonstrated that both bsAb1 and bsAb2 exhibited potent and extensive binding and neutralization efficiencies against multiple SARS-CoV-2 variants including Omicron BA.1-JN.1. Additionally, both bsAbs showed strong potency of inhibiting ACE2 binding. Cryo-electron microscopy (cryo-EM) study revealed detailed interactions at the binding interface, explaining the conservative neutralizing ability of bsAb1 against Omicron variants. The binding sites of H4B6 and H4D12 on the RBD, as well as the differences in these sites, provided insights into the competitive binding observed in the BLI assays. The efficient locking of the entire S protein by bsAb1 highlights a potential mechanism for its superior conservativity and neutralizing effectiveness.
Results
Design and characterization of bsAbs
Given the limitations of monoclonal antibodies in coping with viral immune escape and prominent activity of the IgG-(scFv)2 format [Citation31,Citation32], we engineered two bispecific antibodies using the IgG-(scFv)2 design based on three previously generated human neutralizing antibodies, H4B6, H4D12, and ZW2G10 (a-b). Among them, H4B6 and H4D12 exhibit different neutralizing activities against Omicron subvariants (d), although they target overlapping epitopes on RBD. (h). ZW2G10, an antibody targets RBD and NTD, potently neutralized SARS-CoV-2 and the previously circulating variants of concern (VOCs) Alpha, Beta, Gamma, and Delta, but it failed to neutralize variants containing the R346 T mutation, such as BF.7, XBB.1.5 and XBB.1.16 (d and Supplementary Fig. 1b). We constructed IgG-sc(Fv)2 bsAbs by using the parental antibody with potent activity as the IgG part and the less effective one as the sc(Fv)2 part and linking them using a GGGGSGGGGSGGGG linker [Citation30]. Based on this strategy, we ligated H4B6 and H4D12 to generate bsAb1 (4B6-H-4D12) with overlapping epitopes (a), and linked H4D12 and ZW2G10 to construct another bispecific antibody bsAb2 (4D12-H-2G10) targeting non-overlapping epitopes. Both bsAbs were assembled at the expected molecular weights and showed good purities (c, d). In addition, bsAbs exhibited good thermal stability as verified by differential scanning fluorimetry (e).
Figure 1. Design and characterization of bsAbs. (A–B) Schematic diagram of engineered bispecific antibodies. The bsAb1 was engineered from two parental IgGs H4B6 and H4D12 (A), and the bsAb2 was engineered from two parental IgGs H4D12 and ZW2G10 (B). Fv: variable fragment; scFv: single-chain Fv, constructed as VH-linker-VL; CH1-CL: constant region 1 for heavy chain (CH1) and constant region for light chain (CL); CL-CH1: the crossover format of CH1-CL. (C) Non-reduced and reduced SDS-PAGE showing the size of the bispecific antibodies and their parental antibodies. (D) Characterization of purified antibodies by HPLC-SEC analysis. (E) The thermostability of indicated antibodies using differential scanning fluorimetry (DSF). (F-G) Competitive binding of parental mAbs and bsAbs. Biotinylated spike protein of BA.4/5 was immobilized on streptavidin sensors and then saturated with parental mAbs, and then flowed with bsAbs or parental mAbs. (H) Heatmap showing the competition matrix of parental mAbs and bsAbs. The numbers in the boxes indicate the competition value, i.e. the percentage binding of competing antibody after the binding of primary antibody compared with the binding of competing antibody in the absence of primary antibody. The antibodies were considered fully competitive if the competition value was less than 30% (black box, white numbers), and the antibodies were regarded as partial competition if the competition value was between 30% and 70% (white box, black numbers). If the competition value was greater than 70% (white box, red numbers), the antibodies were judged to noncompete for the same site.
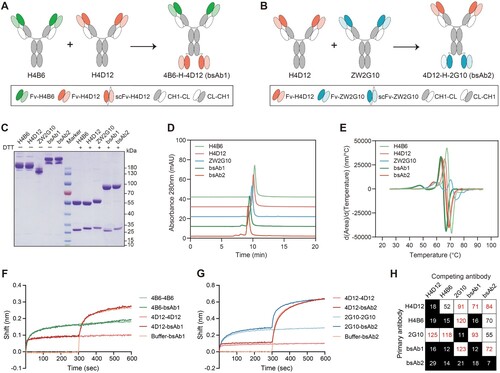
Figure 2. Enhanced neutralizing potency and breadth for bsAbs. (A-C) Neutralization curves of parental mAbs and bsAbs against pseudotyped viruses of Omicron sublineages BA.4/5 (A), BF.7 (B), XBB.1.5 (C). The data were representative of one of at least two independent experiments and were displayed as the mean ± SD. (D) Summary of the IC50 values for pseudotyped viruses determined by nonlinear regression, i.e. log (inhibitor) vs. response (three parameters). (E) Cytopathic effect (CPE) assay of parental mAbs and bsAbs against authentic SARS-CoV-2 WT stain and Omicron subvariants. The IC50 values were calculated by the Reed-Muench method. The cutoff was set at 0.1 and 100 μg/mL (grey bar). Each dot represented one from two or three independent experiments in which four replicates per antibody were performed.
To test the competitive binding ability of parental mAbs and bsAbs to S protein, we performed a competition assay by biolayer interferometry (BLI). The BA.4/5-S protein was biotinylated and immobilized on the streptavidin sensor, which was subsequently saturated by the parental monoclonal antibody (mAb). Then, bsAb was added to the sensor. We found that saturating S with H4D12 first did not affect the binding of bsAb1, instead, pre-binding of the S protein by H4B6 abolished subsequent binding of bsAb1, suggesting that two antigen-binding sites of bsAb1 are similar and the affinity of H4B6 for BA.4/5-S may be stronger than that of H4D12 (f). Furthermore, we found that bsAb2 could still bind to the BA.4/5-S which was first saturated with the parental mAb (g). In contrast, bsAb2 bound to the S protein and blocked the subsequent binding of H4D12 or ZW2G10, suggesting that bsAb2 block two different spike epitopes simultaneously as a single molecule (h). These results indicated that both bsAbs were successfully engineered with bsAb1 and bsAb2 recognizing overlapping and non-overlapping epitopes on the S protein, respectively.
Enhanced neutralizing potency and breadth for bsAbs
To compare the neutralization properties of the bsAbs with parental antibodies and their cocktail, we performed neutralization assays with pseudotyped SARS-CoV-2 (WT) as well as twelve Omicron subvariants BA.1, BA.2, BA.2.75, BA.2.76, BA.4/5, BA.4.6, BF.7, XBB.1.5, XBB.1.16, XBB.2.3, BA.2.86, JN.1 and Delta. H4B6 could effectively neutralize WT, Delta, BA.4/5, BA.4.6, and BF.7 with IC50 values between 18 and 71 ng/mL, but it exhibited reduced neutralizing activity against BA.1, BA.2, BA.2.75, BA.2.76, XBB.1.5, and XBB.1.16 with IC50 values between 101 and 728 ng/mL (d). Unlike the H4B6, H4D12 retained potent neutralization activity against Delta, BA.1, BA.2, BA.2.75, and BA.2.76 but dropped more than 1000-fold against BA.4/5, which evolved from BA.2 (a, d, Supplementary Fig. 1a and Table. 1). Among these pseudoviruses, some have R346 T mutation, dramatically or even completely escaping neutralization of ZW2G10, such as BF.7, which has only one more R346 T mutation than BA.4 in the S protein (b-d and Supplementary Fig. 1b). Different from parental antibodies, bsAbs displayed broad-spectrum neutralization to a range of circulating Omicron subvariants (a-d and Supplementary Table. 1). Remarkably, bsAb1 remained potent in neutralizing Delta and all the Omicron sublineages we tested with IC50 values lower than 20 ng/mL, and it was more potent than its corresponding antibody cocktail (H4B6 + H4D12) in neutralization against BA.4/5 (13-fold), BF.7 (25-fold), XBB.1.5 (102-fold), XBB.1.16 (90-fold), XBB.2.3 (57-fold), BA.2.86 (19-fold) and JN.1 (27-fold) (d and Supplementary Table. 1). Meanwhile, both bsAb1 and bsAb2 showed a slight increase (1.5–12-fold) in neutralization activity against BA.1, BA.2, BA.2.75, and BA.2.76 (d) than that of the cocktail. Moreover, the bsAb2 was significantly superior in neutralizing Omicron BA.4/5, BA.4.6, BF.7, XBB.1.5, XBB.1.16, XBB.2.3, BA.2.86, and JN.1 (IC50 from 1 ng/mL to 25 ng/mL) compared to the ZWD12 + ZW2G10 cocktail (IC50 from 343 ng/mL to over 10 μg/mL) (d and Supplementary Table. 1). For the currently predominantly prevalent JN.1 subvariant, despite bsAb1 binding two overlapping epitopes, it showed better (over 1.4-fold) neutralizing activity than bsAb2.
Next, we evaluated the neutralizing activities of both bsAb1 and bsAb2 against authentic SARS-CoV-2 and four Omicron subvariants using a cytopathic effect (CPE) assay. The bsAb1 effectively neutralized all the tested variants with the IC50 values between 0.11 and 0.49 μg/mL, showing more potent in neutralizing BA.4/5, XBB.1.5, XBB.1.16, and XBB.1.9.2 compared to H4B6 or H4D12 alone (e and Supplementary Table. 1). Compared to bsAb1, bsAb2 exhibited slightly lower neutralization potencies against all tested authentic Omicron subvariants, with IC50 values from 0.28 μg/mL to 0.72 μg/mL, which displayed substantially lower values than that of parental mAbs (e and Supplementary Table. 1).
Consistent with the neutralization results, both bsAbs efficiently bound to spike proteins of various Omicron subvariants, whereas the parental antibodies barely bound to the spike proteins of some of these subvariants (Supplementary Fig. 2a-c and Table. 1). Taken together, these data indicated that bispecific antibodies composed of either non-competing nAbs or competing nAbs possessed a broad synergetic neutralizing capability by constructing two different monoclonal antibodies onto a single molecule.
Antigen-binding properties of bsAbs
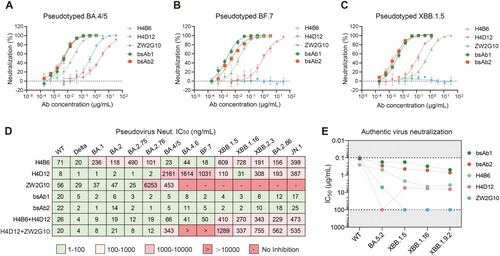
To characterize the binding affinities of the bsAbs and their parental antibodies, we determined the binding kinetics using BLI. We immobilized antibodies onto anti-human IgG Fc (HFc) biosensors and selected spike ectodomain trimers of three Omicron subvariants BA.4/5, BF.7, and XBB.1.5 as analytes to compare the differences between the affinities of the antibodies. The analysis revealed that both bsAbs displayed high affinities to all spike proteins we tested with KD values in the low nanomolar level (a and Supplementary Table. 1). BsAb1 showed an increase of affinity binding to BA.4/5 than H4B6 (2.0-fold change) and H4D12 (8.8-fold change). Notably, H4B6 exhibited 4.5-fold higher affinity than H4D12 to BA.4/5, which might explain why there was a competition between H4B6 and H4D12, but pre-saturation of S with H4D12 did not abolish the subsequent binding of H4B6 (e and a). Besides, bsAb1 bound to the XBB.1.5 spike trimer with a stronger affinity than that of its parental antibodies (a, b, Supplementary Fig. 3 and Table. 1). As we expected, the other bispecific antibody bsAb2 presented strong binding affinities to BA.4/5 (KD = 0.16 nM), BF.7 (KD = 6.54 nM), and XBB.1.5 (KD = 1.27 nM), respectively, whereas ZW2G10 showed no binding affinities to the BF.7 and XBB.1.5 (a, b, Supplementary Fig. 3 and Table. 1). Notably, for BF.7, the affinity of bsAb1 was 4-fold higher than that of bsAb2, which may be due to the fact that, unlike bsAb2, the two parental monoclonal antibodies that compose bsAb1 are insensitive to the R346 T mutation, despite overlapping epitopes. Both bispecific antibodies prevented ACE2 protein binding to RBD in the BLI competition assay, while one parental antibody ZW2G10 did not completely block the S/ACE2 interaction (b).
Figure 3. Antigen-binding properties of bsAbs. (A) Binding kinetics (KD values) of bsAbs and their parental antibodies to the SECD of BA.5, BF.7, and XBB.1.5 as determined using BLI. (B) Kon and Koff of bsAbs and their parental antibodies to the SECD of BA.5, BF.7, and XBB.1.5 as determined by BLI. (C) Antibody blocking of RBD to soluble human ACE2 was detected by BLI. Immobilized biotinylated BA.4/5 RBD were saturated with 300 nM antibody and then flowed through the corresponding antibody in the presence or absence of 300 nM soluble ACE2. As a control, immobilized biotinylated RBD were flowed with PBST buffer and then flowed through an equimolar amount of ACE2. The binding pattern after antibody saturation is shown.
Molecular mechanism of conservative neutralization by bsAb1 and bsAb2
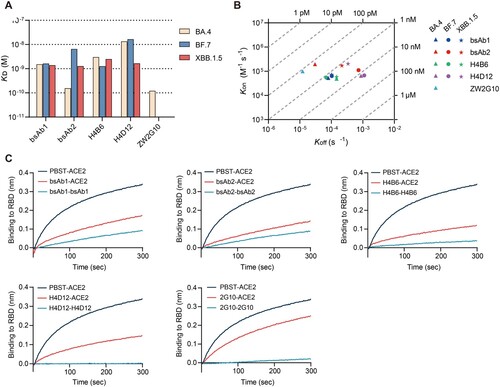
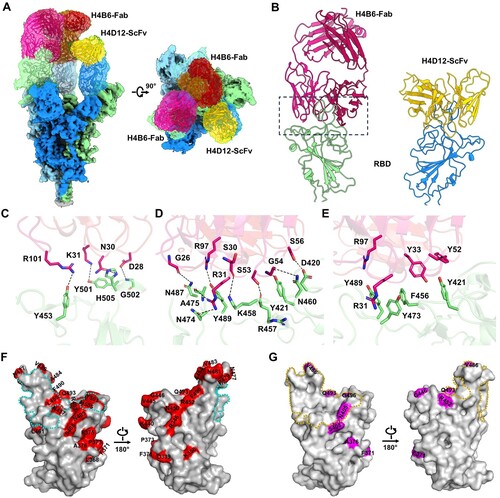
To investigate the molecular mechanism of the conservative neutralizing ability of bsAb1 and bsAb2 against Omicron variants, we incubated these bispecific antibodies with the S protein of the BA.5 variant and subsequently purified the complexes. The cryo-EM structure of the bsAb1-Omicron BA.5 S complex was resolved at a resolution of 2.64 Å (Supplementary Fig. 4 and 5). Notably, all three RBDs of the S protein exhibited an “up” conformation, with a sequential binding of two Fab fragments and one scFv fragment, as inferred from the shape of the density map (a). To enhance the map quality at the binding interface between the RBD domain and H4B6 Fab, we refined the local resolution to 3.4 Å, allowing for detailed modelling of the complex (a, Supplementary Fig. 4 and 5). Both the heavy and light chains of H4B6 Fab primarily participated in binding to RBD, engaging through CDRH1 (residues 25 to 33), CDRH2 (residues 52 to 56), CDRH3 (residues 98 to 105), and CDRL1 (residues 25 to 32) loops (b). The interface between RBD and H4B6 was dominated by a hydrophilic interaction network, forming three clusters. In cluster 1, interactions between D28, N30, and K31 of CDRL1 with Y501, G502, and H505, along with R101 of CDRH3, form hydrogen bonds (c). Cluster 2 includes interactions between S53, G54, and S56 with D420, Y421, and R457. Cluster 3, the largest interface, involve G26, S30, R31, and Y33 of CDRH1, along with R97 of CDRH3, interacting with N487, K458, Y473, A475, L455, and Y489 (d). RBD Y421 and F456 form π-π interactions with Y33 and Y52 of H4B6-H. The cation-π interaction between Y473 of RBD with R31 of H4B6-H stabilizes the interface (e). The identified epitopes on RBD of the S protein almost entirely avoided mutation sites observed in mainstream strains (Alpha, Delta and Omicron, etc.), with only two overlapping residues, N501Y and Y505H (f). These overlaps did not seem to impact the interaction. Furthermore, the L455S mutation in JN.1 relative to BA.5 at the interface potentially increased electrostatic complementarity, aligning with the broad neutralizing capability of H4B6 (f). Unfortunately, we only obtained a low-resolution electron density resembling the binding of RBD to H4D12 scFv. We docked the structural models of H4D12 scFv and RBD into their corresponding region in the cryo-EM map (a, b). Structural analysis reveals that the binding region between H4D12 scFv and the RBD overlaps significantly with that of H4B6 Fab, providing an explanation for the observed competition in the competitive binding assay experiments (Supplementary Fig. 6). Notably, the binding region of H4D12 scFv on the RBD coincides with the three mutation sites of BA.5 relative to BA.1 g), which may explain the higher neutralization efficiency of H4D12 against BA.1 compared to its efficacy against BA.5, surpassing the latter by a remarkable 2000-fold. Upon addition of bsAb2 to the S protein, immediate precipitation of the mixture was observed, impeding the determination of its structural details. However, this propensity to induce antigen aggregation, as evidenced by bsAb2, may underpin its potent neutralizing efficacy.
Figure 4. Cryo-EM structure of the SARS-CoV-2 BA.5 S protein in complex with bsAb1. (A) The domain-coloured cryo-EM structures of the SARS-CoV-2 S protein in complex with bsAb1, viewed along two perpendicular orientations. The H4B6-Fabs and H4D12-ScFv of bsAb1 are coloured magentas, hot pink, and gold, respectively. The three protomers of the S protein are coloured pale green, cyans, and marine. (B) The subcomplex of SARS-CoV-2 RBD with H4B6-Fab or H4D12-ScFv. (C-E) Binding interfaces between RBD and H4B6-Fab. (F) Critical mutation sites (red) associated with mainstream strains on the BA.5 RBD and the binding region of H4B6 Fab (cyan dashed line). (G) The mutation site of BA.5 RBD relative to BA.1 (purple) and the binding region of H4D12 on BA.5 RBD (gold dashed line).
Discussion
The emergence of new variants of SARS-CoV-2 that compromise vaccine efficacy and resist many existing monoclonal antibodies or cocktails, including some currently in clinical trials [Citation21,Citation23–27], highlights the need to develop new strategies against these variants. Rationalization of different mAbs into a single molecule to avoid viral escape under immune stress is a promising therapeutic strategy. In this study, we typically select the non-competitive H4D12 and ZW2G10 to construct the bispecific antibody bsAb2, while a modification was performed to construct the competing H4B6 and H412 into another bispecific antibody bsAb1. In comparison to individual monoclonal antibodies (H4B6 and H4D12), bsAb1 displayed enhanced conservativity and neutralizing effectiveness. This superiority could be attributed to each antibody in bsAb1 possessing four binding interfaces, allowing for better coverage of the entire S protein trimer interface. Based on the resolved structures and the absence of cloudiness observed in bsAb1, we speculate that the bispecific antibody may potentially lock the entire S protein trimer in a monomeric state to inhibit viral invasion.
In summary, the designed bispecific antibodies, bsAb1 and bsAb2, demonstrated superior neutralization capabilities against Delta and a broad range of Omicron subvariants, highlighting their potential as promising therapeutic agents against evolving SARS-CoV-2 variants. In particular, bsAb1, constructed from a non-classical design concept, exhibited similar neutralizing activity to conventional bsAb2 with non-overlapping epitopes through a unique synergistic mechanism, suggesting potential for competitive antibodies in antibody modification.
The findings presented in this study contribute to the growing body of knowledge on the development of bispecific antibodies as potent therapeutics against evolving SARS-CoV-2 variants. The broad-spectrum neutralization observed in bsAbs, coupled with their unique binding profiles and structural characteristics, positions them as promising candidates for further clinical development. As the virus continues to evolve, the adaptability and synergistic neutralization capabilities of bispecific antibodies become increasingly crucial for developing effective therapeutic interventions.
In conclusion, our study demonstrates the successful design and characterization of bispecific antibodies with enhanced neutralizing potency against a diverse range of SARS-CoV-2 variants. The detailed structural insights into the binding interfaces and mechanisms of action provide a foundation for future developments in the field of antibody-based therapeutics, particularly in the context of emerging viral variants. Further investigations, including in vivo studies and clinical trials, will be essential to validate the therapeutic potential of these bispecific antibodies and their applicability in combating the ongoing challenges posed by SARS-CoV-2 and its variants.
Materials and methods
Production of parental mAbs
H4B6 (Chinese Patent Application No. CN202211734994.0), H4D12 (Chinese Patent Application No. CN202211732654.4), and ZW2G10 (Chinese Patent No. ZL202210023619.9) were obtained from donors who were vaccinated with the aerosolized adenovirus type-5 vector-based COVID-19 vaccine (Ad5-nCoV) as described previously [Citation35]. Briefly, we sorted S-specific memory B cells from peripheral blood mononuclear cells (PBMCs) isolated from the whole blood of two donors, and amplified variable regions of the antibody (VH and VL) genes through single-cell PCR technology. Heavy and light chains variable region genes were fused into linearized pcDNA3.4 vectors containing the constant region of the human IgG to construct recombinant expression plasmids. The plasmids of paired heavy and light chain genes were cotransfected into the Expi293F cells (Thermo Fisher Scientific) to express parental monoclonal antibodies. Parental mAbs were purified from cell culture supernatants using the Protein G resin (Cytiva).
Engineering and production of bsAbs
The VH and VL were linked with the GGGGSGGGGSGGGG linker to construct the scFvs as previously described [Citation31]. For engineering bsAb1 (4B6-H-4D12), the scFvs of H4D12 were fused to the C-terminus of H4B6 heavy chain with the same linker to generate modified bsAb1 heavy chain plasmid. The bsAb1 was expressed by co-transfection of the bsAb1 heavy chain and the H4B6 light chain plasmids into Expi293F cells (Thermo Fisher Scientific). Similarly, we constructed another bispecific antibody bsAb2 (4D12-H-2G10) heavy chain plasmid by connecting the scFvs of ZW2G10 to the C terminal of H4D12 heavy chain, and then we co-transfected this plasmid with the H4D12 light chain plasmid at 1:1 ratio into Expi29F cells to generate bsAb2. After 5 days of culture, culture supernatants containing antibodies were harvested and purified using the Protein G resin (Cytiva). Purified bispecific antibodies were concentrated by using ultra centrifugal filters (50 kD; Millipore) and stored in PBS at −80 °C.
Size exclusion chromatography (SEC)-HPLC
For the SEC-HPLC analysis, purified antibodies were subjected to HPLC system (Waters) using a TSKgel G3000SWXL, 5 μm, (7.8 mm × 300 mm) column (TOSOH), which was equilibrated in PBS, pH 7.4. About 10 μg of each antibody in a volume of 20 μL was used for each loading. SEC chromatograms were observed at 280 nm absorbance, with a flow rate of 0.8 mL/min.
Non-reduced and reduced SDS-PAGE
The size and purity of bispecific antibodies were analyzed by SDS-PAGE and Coomassie staining. For reduced SDS-PAGE, 6× protein loading buffer K489 (TransGen) was mixed with 7 μg of each antibody. Samples were loaded on a 4%–12% SDS gradient gel (GenScript) following heating for 5 min at 100 °C. For non-reduced SDS-PAGE, antibodies were mixed with Trident 6× Laemmli SDS sample buffer (Gene Tex) and then loaded directly onto the gel. The gel was run at 180 V for about 50 min and Coomassie staining was performed.
Thermostability analysis
The melting temperature of each antibody was determined through differential scanning fluorimetry (DSF) using UNcle Client software. Briefly, dilute 5000 × SYPRO Orange (Thermo Fisher Scientific) to 500 × with PBS. 48 μL of a 1 mg/ml sample was mixed with 2 μL of diluted SYPRO orange. 9 μl of mixture was added to the Uni tube, and then was heated from 25 °C to 95 °C at a rate of 0.3 °C / min. The fluorescent signal was excited at 473 nm. The area under the fluorescence intensity curve at 510–680 nm, d(Area)/d(Temperature) curve, and primer melting temperature (Tm) values were obtained from the UNcle analysis software.
Enzyme-linked immunosorbent assay
Spike trimer proteins of multiple Omicron subvariants we used were purchased from Acro Biosystems, including BA.1 (SPN-C522a), BA.2 (SPN-C522b), BA.2.75 (SPN-C522f), BA.2.76 (SPN-C522i), BA.3 (SPN-C522c), BA.4/5 (SPN-C5229), BA.4.6 (SPN-C522 m), BF.7 (SPN-C522q), XBB (SPN-C5248), and XBB.1.5 (SPN-C524i). 100 μL 0.2 μg/mL antigen protein was coated into the wells of ELISA microplates per well and incubated overnight at 4°C. After washing with PBS containing 0.2% Tween-20 (PBST), plates were blocked with 2% BSA in PBST for 1 h at 37°C. Following washing with PBST, plates were incubated with fourfold serially diluted antibodies at a 1 μg/mL starting concentration in a 37°C incubator for 1 h and then washed three times with PBST buffer. The bound antibodies were detected by 1:10000 diluted horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Abcam) for 1 h at 37°C. After three washing steps, the plates were incubated by the TMB substrate solution for 6 min at room temperature, and the reaction was quenched by the addition of 2 M H2SO4. The absorbance values were determined at 450 nm with a reference wavelength of 630 nm. Binding curves were fitted using four-parameter nonlinear regression analysis and EC50 values of antibodies were calculated using GraphPad Prism 9 software (version 9.5.1).
BLI-based competitive binding assay
The spike trimer of BA.4/5 was biotinylated using EZ-Link Sulfo-NHS-LC-LC- Biotin (Thermo Fisher Scientific), and then was diluted to a concentration of 50 nM in a running Buffer (PBS containing 0.02% Tween 20 and 0.2% BSA). A biolayer interferometry (BLI) – based competition-binding assay was performed on the GatorPrime BLI System. SA biosensors (Gator) were dipped for 10 min in the running buffer, followed by a baseline signal measurement for 60 s. The biotinylated S protein was immobilized onto SA biosensors for 160 s at 400 rpm. After a wash step in running Buffer for 60 s, the antigen-containing biosensors were immersed into the primary antibody (200 nM) diluted in the same buffer for 300 s at 1000 rpm for the formation of the antibody–antigen complex, followed by incubation with the secondary antibody (200 nM) for 300 s at 1000 rpm for competition binding. The signal of each antibody was normalized with a buffer-only control. The binding signal of competitive antibody was determined as the value of the maximum signal minus the minimum signal at the time of the second antibody contact. The competition value was defined as the percentage of the binding signal of the tested group compared to the binding signal of the control group using buffer as the first antibody. Gator’s software was used to export data, and the binding profile was processed by GraphPad prism 9 Software.
Pseudovirus construction
Genes encoding the full-length spike proteins of SARS-CoV-2 Wuhan Hu-1 (QHD43416.1), Delta (EPI_ISL_2029113), or Omicron/BA.1 (EPI_ISL_12422410) were human codon-optimized and inserted into the pCAGGS vector to construct spike protein-expressing plasmids for the corresponding viral strain. HEK293 T cells were inoculated into cell dishes and cultured overnight at 37 °C and 5% CO2. Until the confluency for adherent cells reached 70–90%, spike protein-expressing plasmids were cotransfected with the HIV backbone vector pNL4-3.Luc.R-E- into HEK293 T cells using the Turbofect transfection reagent (Thermo Fisher Scientific). Supernatants containing pseudotyped virus particles were collected at 48 , 60 , and 72 h post-transfection, filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. Pseudoviruses of other Omicron subvariants were purchased from Vazyme and also used HIV-1 carrying luciferase reporter gene as the viral backbone.
Pseudotyped virus-neutralizing assay
For the neutralization assay, 50 μL of 3-fold serially diluted antibody was mixed and incubated with the same volume of pseudovirus (∼5 × 105 relative luciferase units/well) in 96-well plate at 37 °C for 1 h. 2.5 × 104 ACE2-293 T cells in 100 μL of DMEM supplemented with 10% FBS were subsequently added to the mixture and cultured for 48 h at 37 °C and 5% CO2. Cell controls (without virus and antibodies) and virus controls (with virus only) were set up. 48 h later, luciferase activities were measured using a luciferase assay system (Vazyme) on a TECAN Spark multifunctional microplate detector. The percent neutralization of antibodies was calculated as 100%− (sample signals − cell control signals) / (virus control signals − cell control signals) × 100%. A three-parameter nonlinear regression analysis was performed and IC50 of antibodies was calculated using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA). All data are presented as the means ± standard deviations (SDs).
Authentic virus neutralization CPE assay
Neutralization assay for authentic SARS-CoV-2 and Omicron subvariants using a cytopathic effect (CPE) assay. In detail, 50 μL bispecific antibodies or parental monoclonal antibodies were 2-fold serially diluted in DMEM, with 4 replicate wells for each dilution, mixed with the same volume of the virus (100 CCID50) in 96-well microwell plates, and incubated for 2 h in a 37℃ incubator with 5% CO2. Then, 100 μL cell suspensions of Vero E6 cells (1 × 105 cells/mL) were added to the mixtures and cultured for 5 days. After that, all wells were observed under a microscope and the CPE effect of each well was recorded. The IC50 of each tested sample against different variants was calculated by the Reed-Muench method [Citation36,Citation37]. logIC50 = log (concentration of antibody with > 50% inhibition) + (A-50%)/(A-B) × (−log (dilution factor)). A: Percentage above 50% inhibition; B: Percentage below 50% inhibition. All experiments were performed in a biosafety level 3 (BSL-3) facility.
Affinity determination by BLI
The kinetics of antibodies binding to S protein were performed using a Gator system. After a baseline for 60 s, 50 nM antibodies were used as ligands and immobilized onto anti-human IgG Fc (HFc) biosensors for 100 s at 400 rpm. Following a 60-s stabilization step with running buffer (PBS containing 0.02% Tween 20 and 0.2% BSA), the loaded biosensors were moved into twofold serially diluted (3.125 –100 nM) spike trimer for the 300-s association step. The sensors were then dipped into running buffer for 300 s to detect dissociation. Reference well without antigen was set to correct the background. The curves were fittd using the global fitting method with a 1:1 binding model. Affinity values, including association rates (Kon), dissociation rates (Koff), and affinity constants (KD), were determined with R2 values of greater than 95% confidence level.
hACE2 competition assay
To determine the ability of antibodies to inhibit binding of RBD to ACE2, biotinylated BA.4 RBD was loaded onto streptavidin biosensors for 50 s to reach ∼1 nm binding shift. 300 nM antibodies flowed through the sensor for 300 s, and then sensors were immersed into wells containing the corresponding antibody in the presence of or without 300 nM soluble human ACE2 (Sino) for another 300 s. As a control, immobilized biotinylated RBD were flowed with PBST (PBS containing 0.0.2% Tween) buffer and then flowed through the equal molar of ACE2. The interfering signals from the biotinylated RBD with buffer-only were deducted, and the corrected data were used to compare the competitive characteristics by Octet data analysis software.
Protein preparation
The extracellular domain (ECD) (1-1207 a.a.) of S protein of SARS-CoV-2 (Omicron BA.5 was cloned into the pCAG vector (Invitrogen) with six proline substitutions at residues 815, 890, 897, 940, 984 and 985 and a C-terminal T4 fibritin trimerization motif followed by one Flag tag and a 10xHis tag. This construct will hereafter be referred to as S. A “GSAS” mutation at residues 680 to 683 was introduced into S to prevent the host furin protease digestion. The S protein was purified as below: The recombinant protein was overexpressed using the HEK293F mammalian cells (Invitrogen) at 37℃ under 5% CO2 in a Multitron-Pro shaker (Infors, 130 rpm). When the cell density reached 2.0 × 106 cells/mL, the plasmid was transiently transfected into the cells. To transfect one litre of cell culture, about 1.5 mg of the plasmid was premixed with 3 mg of polyethylenimines (PEIs) (Polysciences) in 50 mL of fresh medium for 30 mins before adding to cell culture. Cells were removed by centrifugation at 4000×g for 15 mins after seventy hours transfection. The secreted proteins were purified by Ni-NTA Agarose (GE Healthcare). After loading two times, the Ni-NTA Agarose was washed with the wash buffer containing 25 mM Tris (pH 8.0), 150 mM NaCl, 50 mM imidazole. The protein was eluted with the elute buffer containing 25 mM Tris (pH 8.0), 150 mM NaCl, 500 mM imidazole. The eluent of S was subject to size exclusion chromatography (Superose 6 Increase 10/300 GL, GE Healthcare) in buffer containing 25 mM Tris (pH 8.0), 150 mM NaCl, and the peak fractions were collected for generation of S -Antibody complexes. The S was incubated with each antibody (baAb1 or bsAb2) at a molar ratio of about 1: 3 for one hour. Then the mixture was subject to size-exclusion chromatography (Superose 6 Increase 10/300 GL, GE Healthcare) in buffer containing 25 mM Tris (pH 8.0), 150 mM NaCl. The peak fractions were collected and concentrated for EM analysis.
Cryo-EM sample preparation and data acquisition
S-bsAb1 complex were concentrated to ∼3.0 mg/mL and applied to the grids, respectively. Aliquots (3.5 μL) of the protein were placed on glow-discharged holey carbon grids (Quantifoil Au R1.2/1.3). The grids were blotted for 3.5 s and flash-frozen in liquid ethane cooled by liquid nitrogen with Vitrobot (Mark IV, Thermo Scientific). The prepared grids were transferred to a Titan Krios operating at 300 kV equipped with a Gatan K3 detector and GIF Quantum energy filter. Movie stacks were automatically collected using EPU software (Thermo Fisher Scientific), with a slit width of 20 eV on the energy filter and a defocus range from –1.2 µm to –2.2 µm in super-resolution mode at a nominal magnification of 81,000×. Each stack was exposed for 2.56 s with an exposure time of 0.08 s per frame, resulting in a total of 32 frames per stack. The total dose rate was ∼50 e-/Å2 for each stack.
Data processing
The movie stacks were motion-corrected with MotionCor2 [Citation38] and binned twofold, resulting in a pixel size of 1.087 Å. Meanwhile, dose weighting was performed [Citation39]. The defocus values were estimated with Gctf [Citation40]. Particles of S alone and S-bsAb1 complex were automatically picked using cryoSPARC [Citation41]. After 2D classification, the particles with clear secondary structure features were selected and subjected to ab-initio reconstruction to obtain the initial models, then multi-hetero refinement without symmetry were performed to selected good particles using cryoSPARC [Citation41]. The selected particles were subjected to non-uniform refinement, local CTF refinement and local refinement with C1 symmetry, resulting in the 3D reconstruction for the whole structures, which was further subject to local refinement with an adapted mask on the interface between RBD of S and bsAb1 to improve the map quality on RBD-bsAb1 subcomplex. The resolution was estimated with the gold-standard Fourier shell correlation 0.143 criterion [Citation42] with high-resolution noise substitution [Citation43]. Refer to Supplementary Figs. S1 and Table S2 for details of data collection and processing.
Model building and structure refinement
For the model building of S, the predicted atomic model by Alphafold [Citation44,Citation45] were used as templates, which were molecular dynamics flexible fitted [Citation46] into the whole cryo-EM map and manually adjusted with Coot [Citation47] to obtain the atomic model of S protein. For the S-bsAb1 complex, the model building was accomplished based on the focused refined. Each residue was manually checked with the chemical properties taken into consideration during model building. Several segments, whose corresponding densities were invisible, were not modelled. Structural refinement was performed in Phenix [Citation48] with secondary structure and geometry restraints to prevent overfitting. Statistics associated with data collection, 3D reconstruction and model building were summarized in Supplementary Table S2.
Author contributions
H.S., X.C., C.Y. conceived the study. H.S., X.C., C.Y., G.Z., P.H., P.F. T.F. identified neutralizing antibodies. H.S., G.Z., P.H., Y.C., T.F. engineered, produced, and characterized the bispecific antibodies. H.S., J.L., X.W. performed binding and neutralization studies. H.S., G.Z. performed affinity and receptor blocking analysis. L.X., Y.Z., Q.Z. performed structural studies. C.Y., X.C., Q.Z. supervised the study. Q.Z. acquired the funding. H.S., L.X. wrote the original draft. H.S., L.X., J.L., Q.Z., X.C., C.Y. reviewed and edited the manuscript.
Supplemental Material
Download MS Word (7.5 MB)Acknowledgements
We thank GISAID and associated laboratories and researchers for the shared sequence information.
Disclosure statement
C.Y., X.C., H.S., G.Z., P.H., P.F., T.F. are listed as inventors on pending patent applications for mAbs H4B6, H4D12 and ZW2G10. The other authors declare no competing interests.
Data and code availability
Atomic coordinates and cryo-EM maps (PDB ID: 8YWW, 8YWX; EMDB ID: EMD-39645, EMD-39646, EMD-39647) have been deposited to the Protein Data Bank (http://www.rcsb.org) and the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/), respectively. Correspondence and requests for materials should be addressed to Q.Z. ([email protected]).
Additional information
Funding
References
- Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi:10.1038/s41586-020-2012-7
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and Is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi:10.1016/j.cell.2020.02.052
- Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292. doi:10.1016/j.cell.2020.02.058
- Davies NG, Jarvis CI, Edmunds WJ, et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 2021;593:270–274. doi:10.1038/s41586-021-03426-1
- Zhou D, Dejnirattisai W, Supasa P, et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184:2348–2361.e6. doi:10.1016/j.cell.2021.02.037
- Wang P, Casner RG, Nair MS, et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021;29:747–751. doi:10.1016/j.chom.2021.04.007
- Mlcochova P, Kemp SA, Dhar MS, et al. SARS-CoV-2 B.1.617.2 delta variant replication and immune evasion. Nature. 2021;599:114–119. doi:10.1038/s41586-021-03944-y
- Viana R, Moyo S, Amoako DG, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in Southern Africa. Nature. 2022;603:679–686. doi:10.1038/s41586-022-04411-y
- Dejnirattisai W, Huo J, Zhou D, et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022;185:467–484. doi:10.1016/j.cell.2021.12.046
- Yamasoba D, Kimura I, Nasser H, et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022;185:2103–2115. doi:10.1016/j.cell.2022.04.035
- Hachmann NP, Miller J, Collier AY, et al. Neutralization escape by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4, and BA.5. N Engl J Med. 2022;387:86–88. doi:10.1056/NEJMoa2204705
- Yang S, Yu Y, Xu Y, et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect Dis. 2024;24:e70–e72. doi:10.1016/S1473-3099(23)00744-2
- CDC. COVID Data Tracker. Cent. Dis. Control Prev. 2020. [cited 2024 Feb 26]. Available from: https://covid.cdc.gov/covid-data-tracker.
- Hoffmann M, Krüger N, Schulz S, et al. The Omicron variant is highly resistant against antibody-mediated neutralization: implications for control of the COVID-19 pandemic. Cell. 2022;185:447–456. doi:10.1016/j.cell.2021.12.032
- Planas D, Saunders N, Maes P, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–675. doi:10.1038/s41586-021-04389-z
- Cele S, Jackson L, Khoury DS, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602:654–656. doi:10.1038/s41586-021-04387-1
- Uraki R, Ito M, Kiso M, et al. Antiviral and bivalent vaccine efficacy against an omicron XBB.1.5 isolate. Lancet Infect Dis. 2023;23:402–403. doi:10.1016/S1473-3099(23)00070-1
- Qu P, Evans JP, Faraone JN, et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe. 2023;31:9–17. doi:10.1016/j.chom.2022.11.012
- Wang H, Xue Q, Zhang H, et al. Neutralization against Omicron subvariants after BA.5/BF.7 breakthrough infection weakened as virus evolution and aging despite repeated prototype-based vaccination. Emerg Microbes Infect. 2023;12:2249121. doi:10.1080/22221751.2023.2249121
- Jeworowski LM, Mühlemann B, Walper F, et al. Humoral immune escape by current SARS-CoV-2 variants BA.2.86 and JN.1, December 2023. Euro Surveill. 2024;29:2300740. doi:10.2807/1560-7917.ES.2024.29.2.2300740
- Cao Y, Wang J, Jian F, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022;602:657–663. doi:10.1038/s41586-021-04385-3
- Takashita E, Kinoshita N, Yamayoshi S, et al. Efficacy of antiviral agents against the SARS-CoV-2 Omicron subvariant BA.2. N Engl J Med. 2022;386:1475–1477. doi:10.1056/NEJMc2201933
- Imai M, Ito M, Kiso M, et al. Efficacy of antiviral agents against Omicron subvariants BQ.1.1 and XBB. N Engl J Med. 2023;388:89–91. doi:10.1056/NEJMc2214302
- Iketani S, Liu L, Guo Y, et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature. 2022;604:553–556. doi:10.1038/s41586-022-04594-4
- Cameroni E, Bowen JE, Rosen LE, et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602:664–670. doi:10.1038/s41586-021-04386-2
- Cao Y, Yisimayi A, Jian F, et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022;608:593–602. doi:10.1038/s41586-022-04980-y
- Yue C, Song W, Wang L, et al. ACE2 binding and antibody evasion in enhanced transmissibility of XBB.1.5. Lancet Infect Dis. 2023;23:278–280. doi:10.1016/S1473-3099(23)00010-5
- Chi H, Wang L, Liu C, et al. An engineered IgG–VHH bispecific antibody against SARS-CoV-2 and Its variants. Small Methods. 2022;6:e2200932). doi:10.1002/smtd.202200932
- Wang Y, Zhang X, Ma Y, et al. Combating the SARS-CoV-2 Omicron (BA.1) and BA.2 with potent bispecific antibodies engineered from non-Omicron neutralizing antibodies. Cell Discov. 2022;8:104. doi:10.1038/s41421-022-00463-6
- Cho H, Gonzales-Wartz KK, Huang D, et al. Bispecific antibodies targeting distinct regions of the spike protein potently neutralize SARS-CoV-2 variants of concern. Sci Transl Med. 2021;13:eabj5413. doi:10.1126/scitranslmed.abj5413
- Ku Z, Xie X, Lin J, et al. Engineering SARS-CoV-2 specific cocktail antibodies into a bispecific format improves neutralizing potency and breadth. Nat Commun. 2022;13:5552. doi:10.1038/s41467-022-33284-y
- Li Z, Li S, Zhang G, et al. An engineered bispecific human monoclonal antibody against SARS-CoV-2. Nat Immunol. 2022;23:423–430. doi:10.1038/s41590-022-01138-w
- Liu R, Oldham RJ, Teal E, et al. Fc-Engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies Basel Switz. 2020;9:64. doi:10.3390/antib9040064
- Kubota T, Niwa R, Satoh M, et al. Engineered therapeutic antibodies with improved effector functions. Cancer Sci. 2009;100:1566–1572. doi:10.1111/j.1349-7006.2009.01282.x
- Chi X, Guo Y, Zhang G, et al. Broadly neutralizing antibodies against Omicron-included SARS-CoV-2 variants induced by vaccination. Signal Transduct Target Ther. 2022;7:139. doi:10.1038/s41392-022-00987-z
- Lei C, Yang J, Hu J, et al. On the calculation of TCID50 for quantitation of virus infectivity. Virol Sin. 2021;36:141–144. doi:10.1007/s12250-020-00230-5
- Khoury DS, Wheatley AK, Ramuta MD, et al. Measuring immunity to SARS-CoV-2 infection: comparing assays and animal models. Nat Rev Immunol. 2020;20:727–738. doi:10.1038/s41577-020-00471-1
- Zheng SQ, Palovcak E, Armache J-P, et al. Motioncor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. doi:10.1038/nmeth.4193
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 2015;4:e06980. doi:10.7554/eLife.06980
- Zhang K. Gctf: real-time CTF determination and correction. J Struct Biol. 2016;193:1–12. doi:10.1016/j.jsb.2015.11.003
- Punjani A, Rubinstein JL, Fleet DJ, et al. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 2017;14:290–296. doi:10.1038/nmeth.4169
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi:10.1016/j.jmb.2003.07.013
- Chen S, McMullan G, Faruqi AR, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi:10.1016/j.ultramic.2013.06.004
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi:10.1038/s41586-021-03819-2
- Varadi M, Anyango S, Deshpande M, et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi:10.1093/nar/gkab1061
- Trabuco LG, Villa E, Mitra K, et al. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi:10.1016/j.str.2008.03.005
- Emsley P, Lohkamp B, Scott WG, et al. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi:10.1107/S0907444910007493
- Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi:10.1107/S0907444910026582