
ABSTRACT
Bats have long been associated with multiple pathogens, including viruses affecting humans such as henipaviruses, filoviruses, bunyaviruses and coronaviruses. The alpha and beta coronaviruses genera can infect most mammalian species. Among them, betacoronavirus SARS-CoV, MERS-CoV and SARS-CoV-2, which have caused the three major pandemics in the last two decades, have been proposed to originate in bats. In this study, 194 oral swabs from 22 bats species sampled in 19 locations of the Iberian Peninsula were analysed and characterized by three different PCR tests (coronavirus generic real-time RT-PCR, multiplex conventional PCR, and SARS-CoV-2 specific real-time RT-PCR) to detect bat coronaviruses. Screening with coronavirus generic PCR showed 102 positives out of 194 oral swabs analysed. Then, metabarcoding with multiplex PCR amplified 15 positive samples. Most of the coronaviruses detected in this study belong to alphacoronavirus (α-CoV) genus, with multiple alphacoronaviruses identified by up to five different genetic variants coexisting in the same bat. One of the positive samples identified in a Miniopterus schreibersii bat positive for the generic coronavirus PCR and the specific SARS-CoV-2 PCR was classified as betacoronavirus (β-CoV) through phylogenetic analysis. These results support the rapid evolution of coronaviruses to generate new genomic potentially pathogenic variants likely through co-infection and recombination.
1. Introduction
Chiroptera order is one of the most abundant, diverse, and widespread group of mammals, comprising approximately 1,200 species and representing almost 25% of the class Mammalia. Bats are the second biggest order of mammals and the only ones capable of flying. Thirty-one species of this order are native to the Iberian Peninsula. In recent years, bats have been of particular interest not only for their ecological implications, but also for public health reasons, as they are considered to play an important role in the emergence and transmission of zoonotic pathogens [Citation1]. These animals are vectors and natural reservoirs for a wide range of microorganisms with the ability to cross the species barrier, which makes them potential sources of zoonotic pathogens.
Bats have been associated with highly pathogenic human viruses, acting as hosts for recombination, transmission and spread of these pathogens. Examples include lyssaviruses (rabies virus), henipaviruses (Nipah virus and Hendra virus), filoviruses (Marburg virus, Ebola virus and Mengla virus), bunyaviruses (Ahun virus) [Citation1–3] and coronaviruses, putative precursors of the pandemic-associated viruses SARS-CoV [Citation4–6], MERS-CoV [Citation7–10] and more recently SARS-CoV-2 [Citation11–14]. SARS-CoV emerged in 2002–2003 in the Guangdong province, China, and bats were identified as the reservoir and probable source of this emerging disease-causing pathogen [Citation4,Citation5]. Ten years later, in 2012, MERS-CoV emerged in the Middle East and although in this case camels were identified as the source for human infection, closely related viruses to MERS-CoV were also identified in bats [Citation15]. The last COVID-19 pandemic caused by SARS-CoV-2 probably had its origin in wild animals, and an association between bats and this emerging zoonotic disease has been established [Citation11–14]. For this reason, the surveillance of bats and other wild animals that could act as potential reservoirs for these pathogens has been promoted.
Alphacoronaviruses (α-CoV) and Betacoronaviruses (β-CoV) infect several mammalian species, including humans, bats and pigs, while Gammacoronaviruses (γ-CoV) and Deltacoronaviruses (δ-CoV) infect birds, wild felines, pigs and some marine mammalian species [Citation16,Citation17]. Both α-CoVs and β-CoVs have been identified in bats from different European countries such as Italy [Citation18–24], France [Citation25,Citation26], United Kingdom [Citation27,Citation28], Germany [Citation29–31], Romania [Citation29], the Netherlands [Citation29,Citation32], Ukraine [Citation29], Finland [Citation33], Denmark [Citation34,Citation35], Hungary [Citation36], Bulgaria [Citation37], Slovenia [Citation6], Luxembourg [Citation38] and Switzerland [Citation39]. In some cases, both α-CoVs and β-CoVs genera coexist in the same animal, as it has been detected in the Iberian Peninsula or other locations [Citation40,Citation41].
One of the priorities of the World Health Organization (WHO) is the surveillance of emerging diseases and the development of preventive and control interventions. The WHO gives priority to emerging diseases associated with public health risk due to their epidemic potential and/or limited or insufficient control interventions. Some of the viruses associated with bats (https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts) are associated with these prioritized emerging diseases.
The aim of this study was the detection and classification of coronaviruses present in Iberian bat species to advance knowledge on their prevalence, and to improve control and surveillance measures for potentially new emerging zoonotic coronaviruses.
2. Materials and methods
2.1. Ethical statement
The authors confirm that the ethical policies of the journal have been adhered to. Samples were obtained and processed following standard operating procedures with the appropriate approval of the Ethical and Scientific Committees (AUES/CYL/208bis/2021; AUF/202170016; EB/039/2019-1).
2.2. Sample collection
To detect the presence and distribution of the different coronaviruses in Spanish bats, 194 animals belonging to 22 species were captured with mist nets and harp traps setting up in activity areas (ponds, riverine habitats, etc.) and close to roost [Citation42], at 19 sites distributed by five provinces of Spain during the summer of 2020 (; ). Capture, handling, and sampling of animals were carried out by trained personnel, complying with all relevant national guidelines and institutional policies, and in strict accordance with good animal practices. Moreover, instruments were sterilized between captures, and personnel was protected with gloves and facemasks, following the specific regulations of working with bats in a pandemic scenario, according to the Spanish Association for the Conservation and Study of Bats (www.secemu.org). Each animal was maintained in individual sterilized cotton bags to avoid sample contamination before morphologically identified by expert personnel using taxonomic keys [Citation43], and straightaway oro-pharyngeal swabs were collected; finally, the animals were released.
Table 1. Sample collection distribution and classification.
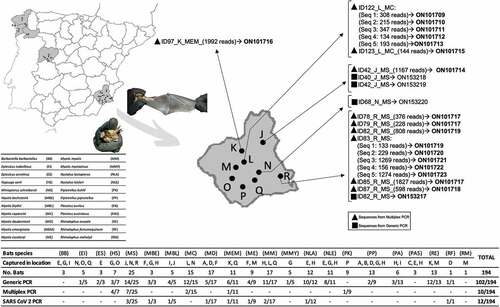
Figure 1. Distribution of captured bat species in the sampled areas showing the results of coronavirus detection.
2.3. RNA extraction
The oropharyngeal swabs were preserved in Tri-Reagent (Sigma-Aldrich, Burlington, MA, USA) at −80°C. RNA extraction was conducted in a biosafety level 3 cabinet. Tri-Reagent was used for nucleic acid extraction according to the manufacturer’s instructions.
2.4. Gene amplification and sequence analyses
Samples were analysed based on the three techniques detailed below.
2.4.1. Generic real-time RT-PCR
A described real-time RT-PCR [Citation44] was user for the generic screening of coronaviruses. The 194 RNA samples were analysed by a real-time RT-PCR for the generic detection of coronaviruses, targeting the most conserved fragment belonging to ORF1b region and amplifying a fragment of 179 bp [Citation44]. Sequences of the primers are described in . All reactions were carried out on a CFX96 Touch Real Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using the iTaq Universal One-Step RT-qPCR kit with SYBR Green (Bio-Rad, Hercules, CA, USA). Volume reaction was adjusted to 15 µl and included 7.5 µl reaction mix, 1 µl of each primer at 10 µM, 0.25 µl of iScript reverse transcriptase, 2 µl of RNA and 3.25 µl of nuclease free water to complete the reaction volume. Conditions of the real-time RT-PCR were as follows: reverse transcription was carried out at 50°C for 15 min, followed by polymerase activation and first denaturation at 95°C for 1 min and by 40 cycles of 95°C for 10 sec, 50°C for 20 sec and 60°C for 20 sec. Melting curve analysis was performed by heating the samples to 95°C at 0.5°C increments every 6 sec.
Table 2. Primers used for partial gene amplifications.
PCR products were sequenced and aligned with reference sequences obtained from GenBank using ClustalW. Reference sequences included different bat coronavirus, as well as SARS-CoV, SARS CoV-2 and MERS-CoV strains isolated from humans. The fragment analysed was 118 bp, and a phylogenetic tree was generated using MEGA version 10 (http://www.megasoftware.net) with the maximum likelihood method with Tamura 3-parameter model [Citation45]. Model of the sequence evolution was selected based on Corrected Akaike Information Criterion (cAIC) and Bayesian Information Criterion (BIC); the model with lowest values of cAIC and BIC was chosen. Bootstrap confidence limits were calculated based on 1000 replicates; branch numbers in the tree indicate bootstrap results ().
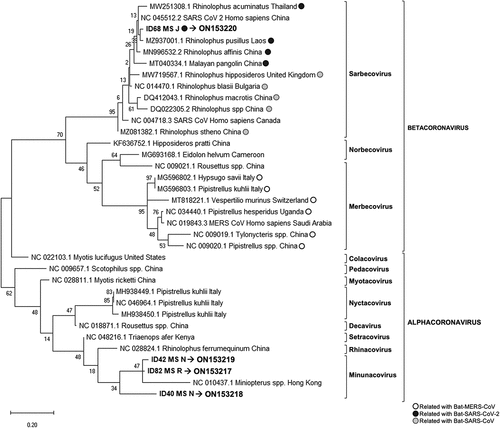
Figure 2. Phylogenetic tree generated for the 118 bp fragment from the coronavirus generic PCR (ORF1b region).
2.4.2. Conventional multiplex PCR
In addition, a novel conventional multiplex PCR designed for this study was performed to characterize and sequence the community of coronaviruses co-infecting a bat, by using a multiplex primer approach and a high-throughput sequencing technology. To design general primers for coronaviruses, 65 genomes available in GenBank for different animal species (list of genomes in Supporting Information) were downloaded and aligned with Clustal X, using the program UGENE (http://ugene.net). Primer sites were selected in two of the most conserved regions of the genomes, amplifying a fragment of 422 bp within the ORF1ab polyprotein gene, between the nucleotide positions 14798 and 15219 of the reference genome sequence NC_048212.1. This region showed a high divergence among genomes in preliminary phylogenetic analysis. A set of 12 primers (7 forward and 5 reverse) were designed to potentially amplify the 65 genomes, following the default conditions of the program UGENE, and ensuring that no more than five degenerated bases were added at each primer to minimize cross amplifications with bat genome (). Reverse transcription was performed with the First-Strand cDNA Synthesis Kit (NZYTech, Lisbon, Portugal) according to the manufacturer’s instructions. PCR reactions were carried out in a T100 Thermal cycler (Bio-Rad, Hercules, CA, USA) using the Multiplex PCR Kit (QIAGEN, Hilden, Germany) and the mix of primers (0.4 µl of each primer at 100 µM in a final volume of 50 µl). Avian and human positive controls (identified as APC and HPC, respectively) were used to check and optimize the PCR conditions. Volume reaction was adjusted at 10 µl, and included 5 µl reaction mix, 1 µl of primer mix, 2 µl of cDNA and 2 µl of nuclease free water to complete the reaction volume. Conditions of the Multiplex-PCR were as follows: 95°C for 15 min, followed by 7 cycles of 95°C for 30 sec, 56°C for 45 sec, decreasing −.5°C per cycle and 72°C for 30 sec, and another 38 cycles of 95°C for 30 sec, 53°C for 1 min and 30 sec, and 72°C for 30 sec, with a final extension of 60°C for 5 min. PCR products were run on an electrophoresis gel and, for samples amplifying a product with the expected size, PCR products were amplified again to incorporate Illumina indexes using the KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cape Town, South Africa) recommended for amplicon library preparation by Illumina. Each PCR product included its own, unique, index sequence. PCR reaction volume was 14 µl and included: 7 µl 2x KAPA HiFi HotStart ReadyMix, 1.4 µl of mixed indexing primer at 10 mM, 2.8 µl of PCR product and 2.8 µl of nuclease free water. Indexing thermal cycling conditions were 95°C for 3 min, followed by 8 cycles of 95°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec, with a final extension of 72°C for 5 min. Index PCR products were visualized by electrophoresis in agarose gel. Positive samples were purified using 0,8X AMPure®XP beads, quantified in Epoch spectrophotometer (Agilent, Santa Clara, CA, USA) and normalized at 20 nM before pooled. Final library was validated in the TapeStation System (Agilent, Santa Clara, CA, USA) using High Sensitivity D1000 Screen Tape assay and purified again using 0,68X AMPure®XP beads to clean up fragments around 250 bp. After this clean up, the pool was quantified in Qubit (ThermoFisher, Waltham, MA, USA) and in the pool concentration was tested by a qPCR using KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems, Cape Town, South Africa). Finally, double indexed PCR amplicons were sequenced in an Illumina MiSeq System using MiSeq V2 500-cycle reagent kit with paired-end reads (Illumina, San Diego, CA, USA). Sequences obtained by NGS were aligned by read pairs, purified, and cleaned using ObiTools software [Citation46] to align and remove any sequence errors, primer sequences and fragments with less than 380 bp and 100 reads. After the first bioinformatic analysis with ObiTools, these sequences were analysed by performing a phylogenetic tree with the same software described above. In this case, the analysed fragment was 381 bp and the phylogenetic tree was generated using the maximum likelihood method with General Time Reversible model [Citation47], which had the lowest values of cAIC and BIC. Bootstrap confidence limits were calculated based on 1000 replicates, branch numbers in the tree indicate Bootstrap results ().
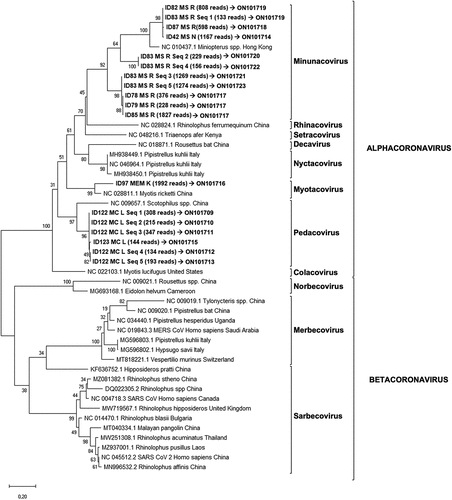
Figure 3. Phylogenetic tree generated for the 381 bp fragment from the coronavirus Multiplex-PCR (ORF1ab region).
2.4.3. SARS-CoV-2 specific real-time RT-PCR
All positive samples in the PCRs described above were analysed using a specific real-time RT-PCR for SARS-CoV-2 developed by the Center for Disease Control and Prevention (CDC). Primers and probes sequences are described in , which were used to individually amplify two targets of virus nucleocapsid gene (N1 and N2). All reactions were carried out on the CFX96 Touch Real-Time PCR Detection System using the SuperScript™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase reagent kit (ThermoFisher, Waltham, MA, USA). Volume reaction was adjusted at 15 µl, and included 7.5 µl of 2X Reaction Mix, 0.32 µl of Magnesium Sulphate (5 mM), 0.8 µl of SuperScriptTM III RT PlatinumTM Taq Mix, 1 µl of mix of primers and probes (10 µM), 2 µl of RNA and 3.38 µl of nuclease free water. Conditions of real-time RT-PCR were as follows: reverse transcription was carried out at 55°C for 20 min, followed by polymerase activation and first denaturation at 95°C for 3 min and by 50 cycles split in two steps: 95°C for 3 sec, 55°C for 30 sec.
3. Results
At least one of the α-CoV variant was detected by generic coronavirus real-time RT-PCR or conventional multiplex PCR in all sampled locations except in one site (location C), while a β-CoV was detected only in one place (location N) (). Nineteen bat species out of 22 sampled were positive for the presence of coronavirus, being Barbastella barbastellus, Plecotus austriacus and Rhinolophus mehelyii the only bat species that showed no positive results ().
The generic real-time RT-PCR revealed 102 samples positives for coronavirus out of the 194, detected in 14 locations and in 18 bat species. Four out of the 102 positive samples were sequenced for the fragment of 118 bp of ORF1b region, and all were isolated from the species M. schreibersii, two from location J, one from location R and one from location N (). These sequences were compared with the sequences available in the GenBank database and showed a homology between 86% and 96%. In the phylogenetic tree, these sequences clustered in two groups, samples with ID: 40, 42, and 82 clustered with α-CoV, and only the sequence from sample ID 68 was clustered with sarbecoviruses, within the β-CoV (). The accession numbers of these new four sequences are ON153217-ON153220.
Conventional PCR revealed 15 samples positive for coronavirus out of 194, detected in seven locations and in five bat species. Five samples were discarded as the coverage of sequences obtained was less than 100 reads. For the remaining 10 samples, 18 distinct viral sequences were obtained, and ranged between 1 and 5 sequences per sample (). One same viral sequence was obtained from eight distinct samples (ID 42, 78, 79, 82, 85, 87, 97 and 123; ), while five different sequences were obtained in the same sample, ID 83 and 122 (). The accession numbers of these sequences are ON101709-ON101723. Sequences from positive samples ID 78, 79 and 85 are homologous to each other (GenBank accession number ON101717). Sequences detected in samples ID 82 and 83 (Seq_1) are also homologous to each other ( & ; GenBank accession number ON101719). All studied sequences were from four locations of the same province, Murcia, belonging to four bat species: Hypsugo savii, M. schreibersii, Myotis capaccini and Pipistrellus kuhlii, and showed homology between 86% and 100% to the sequences from the GenBank database (). In the phylogenetic tree, the 18 sequences clustered in the α-CoV group ().
Table 3. NGS sequencing results.
The SARS-CoV-2 PCR showed 12 positives out of 194 samples. These PCR products were less than 100 bp and the information provided by sequencing of these fragments was limited. Nevertheless, all positive samples for this PCR were also positive in the real-time RT-PCR described above.
4. Discussion
Bats play a key role in both natural and anthropized ecosystems with fundamental contributions to human wellbeing, such as feeding on agricultural pests, pollination, and ecological seed dispersal [Citation48,Citation49]. They also play an important role in the health of these ecosystems and are intrinsically intertwined with animal and human health as approached in the One Health concept [Citation50]. Bats are considered the ancestral hosts of many pathogenic viruses from different families, including lyssaviruses, henipaviruses, filoviruses, bunyaviruses and coronaviruses [Citation51]. Recently, it has been shown that novel coronavirus closely related to SARS-CoV-2 may be transmitted by bats to other species and then passed on to humans [Citation11–14].
Among the 31 known native bat species in the Iberian Peninsula, a total of 22 were sampled from the West to the East of mainland Spain. Of the 194 bats analysed, 53% tested positive for coronaviruses in at least one of the three PCR tests performed. These results suggested a high prevalence of these viruses in bats from the Iberian Peninsula. Viral detection by conventional PCR was lower than that obtained by the generic real-time PCR, possibly due among other reasons, to the different sensitivity of the techniques and size of the amplified fragments. The real-time RT-PCR analysis showed several peaks in the melting curve with differences of 0.5°C between positive samples and the positive control (data not shown), suggesting that different variants of coronavirus were amplified. Indeed, sequencing and phylogenetic analysis revealed that these viruses were classified as α-CoV and β-CoV and, according to the current ICTV classification system for coronaviruses [Citation52], as Minunacovirus, Myotacovirus, Pedacovirus and Sarbecovirus ().
The success of the conventional Sanger sequencing (only 4/102 samples were successfully sequenced) might be affected by the small fragment size, together with the likely amplification of more than one strain from a single sample, suggesting that multiple coronavirus strains are present in the same sample. Additionally, this was further confirmed by the conventional multiplex PCR, where more than one α-CoV variant was obtained from same sample (bats ID 83 and 122) with up to 5 sequence variants detected. Recombination is one of the major drivers of virus evolution; considering the rapid evolution and complexity of these viruses with sufficient capacity to generate new genomes, the coexistence of different coronaviruses in the same bat enables the existence of recombination between them inside the animal to potentially become a risk for other animals and humans [Citation53]. Furthermore, these results confirmed the ability of bats to resist and tolerate viruses with low viraemia and the absence of clinical symptoms [Citation51], which has been observed even in experimentally infected bats with highly lethal viruses such as henipavirus [Citation54,Citation55], MERS-CoV [Citation56], Ebola virus [Citation57], and Marburg virus [Citation58]. However, future cohort and long-term studies must determine the impact of coronaviruses, in terms of infection and evolution of disease, in native bats from the Iberian Peninsula.
Bats live in colonies of a few to thousands of individuals and possess a high longevity compared to other mammals of their size [Citation59]. This, together with their ability to fly, explains why bats are exposed to a wide variety of viruses, which influences the evolution of their immune system making them resistant to different viruses [Citation60]. The first line of defence against any virus is the innate immune response, which is also related to bat flying ability, which requires metabolic adaptation to rapid increases in activity, body temperature and associated molecular damage. Therefore, the ability to fly could be related to bat capacity to regulate the associated inflammatory processes, which would also give them an adaptive advantage in resistance to pathogens [Citation61].
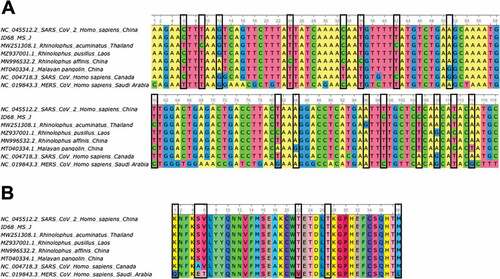
Most of the coronaviruses detected in this study belong to α-CoV genus but one of them was classified as β-CoV through phylogenetic analysis. In this case, the sample ID 68 was positive for two of the molecular tests, the real-time RT-PCR [Citation44] and the specific SARS-CoV-2 PCR (https://www.cdc.gov/coronavirus/2019-ncov/lab/virus-requests.html). Although it was positive for SARS-CoV-2 PCR, it cannot be confirmed since the information provided by this PCR is very limited because it amplifies a short fragment of SARS-CoV-2 genome and thus confirmation would require deepest sequencing of the genome. However, the sequence obtained from the real-time RT-PCR for this sample supported that this is a coronavirus of the β-CoV genus, closely related to SARS-CoV-2 (). Subsequently, the rapid evolution and complexity of these viruses with sufficient capacity to generate new genomes through co-infection and recombination may facilitate cross-species transmission [Citation51,Citation62]. Furthermore, our results provide additional support for the potential risk of SARS-CoV re-emergence from viruses currently circulating in bat populations [Citation63].
Figure 4. A) Alignment of partial ORF1b gene sequences (118 bp) of some bat-related SARS-CoV-2 (MN996532.2, MZ937001.1, MW251308.1) and pangolin (MT040334.1) coronavirus isolates, including the SARS-CoV-2 Wuhan-Hu-1 (NC_045512.2), SARS-CoV (NC_004718.3) and MERS-CoV (NC_019843.3) reference sequences and the sequence obtained in this study ID 68 (ON153220). Nucleotide changes between bat and pangolin isolates related to SARS-CoV-2 with the reference sequence for this virus are highlighted in black boxes. B) Amino acid alignment (39 aa) of partial ORF1b gene sequences (118bp) described in section A of this figure. Amino acid changes between bat and pangolin isolates related to SARS-CoV-2 with the reference sequence for SARS-CoV and MERS-CoV are highlighted in black boxes.
Bats are reservoirs for a wide range of different viruses including coronaviruses where they are putative precursors of pandemic-associated viruses SARS-CoV [Citation4–6], MERS-CoV [Citation7–10] and more recently SARS-CoV-2 [Citation11–14]. The risk of emergence of zoonotic pathogens in humans depends on the interactions between humans and infected animals, reservoirs and/or vectors, or their environment. Knowledge of the causal relationships between the human-animal interface and the emergence of pathogens in humans is still incomplete. However, the climate change, land use change (deforestation, urbanization, crop intensification), habitat alterations, changes in animal production management and in food and water availability, and other human activities favour and increase in the interactions between humans and other animals [Citation64]. As with other viruses prior to the SARS-CoV-2 pandemic, the emergence of numerous viruses has been attributed to increased interspecific contact between humans and wildlife following increased bushmeat hunting and invasion on undisturbed habitats [Citation64,Citation65].
In conclusion, the results of this study encourage research to better understand and characterize coronavirus genetic variability and natural hosts. Extensive viral sampling in multiple bat species and other wild and domestic animals in many different locations, epidemiological and ecological contexts is required to improve control and prevention measures against newly associated emerging zoonotic diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Dacheux L, Cervantes-Gonzalez M, Guigon G, et al. A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses. PloS One. 2014;9(1):e87194.
- Wang LF, Cowled C, Eds. Bats and viruses: a new frontier of emerging infectious diseases. Hoboken, NJ: John Wiley & Sons. 2015.
- Yang XL, Tan CW, Anderson DE, et al. Characterization of a filovirus (Měnglà virus) from Rousettus bats in China. Nat Microbiol. 2019;4(3):390–395.
- Lau SK, Woo PC, Li KS, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA. 2005;102(39):14040–14045.
- Li W, Shi Z, Yu M, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–679.
- Rihtaric D, Hostnik P, Steyer A, et al. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch Virol. 2010;155(4):507–514.
- Woo PC, Lau SK, Li KS, et al. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg Microbes Infect. 2012;1(1):e35.
- Lau SK, Li KS, Tsang AK, et al. Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J Virol. 2013;87(15):8638–8650.
- Corman VM, Ithete NL, Richards LR, et al. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J Virol. 2014;88(19):11297–11303.
- Anthony SJ, Johnson CK, Greig DJ, et al. Global patterns in coronavirus diversity. Virus Evol. 2017;3(1):vex012.
- Zhou H, Chen X, Hu T, et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr Biol. 2020;30(19):3896.
- Zhou H, Ji J, Chen X, et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. 2021;184(17):4380–4391.e14.
- Wacharapluesadee S, Tan CW, Maneeorn P, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat Commun. 2021;12(1):972.
- Temmam S, Vongphayloth K, Baquero E, et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature. 2022;604(7905):330–336.
- Anthony SJ, Gilardi K, Menachery VD, et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. MBio. 2017;8(2):e00373–17.
- Woo PC, Lau SK, Huang Y, et al. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med. 2009;234(10):1117–1127.
- Woo PC, Lau SK, Lam CS, et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995–4008.
- Rizzo F, Edenborough KM, Toffoli R, et al. Coronavirus and paramyxovirus in bats from Northwest Italy. BMC Vet Res. 2017;13(1):396.
- de Sabato L, Lelli D, Faccin F, et al. Full genome characterization of two novel Alpha-coronavirus species from Italian bats. Virus Res. 2019;260:60–66.
- Lecis R, Mucedda M, Pidinchedda E, et al. Molecular identification of Betacoronavirus in bats from Sardinia (Italy): first detection and phylogeny. Virus Genes. 2019;55(1):60–67.
- Lelli D, Papetti A, Sabelli C, et al. Detection of coronaviruses in bats of various species in Italy. Viruses. 2013;5(11):2679–2689.
- de Benedictis P, Marciano S, Scaravelli D, et al. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes. 2014;48(2):366–371.
- Moreno A, Lelli D, de Sabato L, et al. Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy. Virol J. 2017;14(1):239.
- Balboni A, Palladini A, Bogliani G, et al. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiol Infect. 2011;139(2):216–219.
- Monchatre-Leroy E, Boué F, Boucher JM, et al. Identification of alpha and beta coronavirus in wildlife species in France: bats, rodents, rabbits, and hedgehogs. Viruses. 2017;9(12):364.
- Goffard A, Demanche C, Arthur L, et al. Alphacoronaviruses detected in French bats are phylogeographically linked to coronaviruses of European Bats. Viruses. 2015;7(12):6279–6290.
- Crook JM, Murphy I, Carter DP, et al. Metagenomic identification of a new sarbecovirus from horseshoe bats in Europe. Sci Rep. 2021;11(1):14723.
- August TA, Mathews F, Nunn MA. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 2012;12(6):530–533.
- Annan A, Baldwin HJ, Corman VM, et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg Infect Dis. 2013;19(3):456–459.
- Gloza-Rausch F, Ipsen A, Seebens A, et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14(4):626–631.
- Fischer K, Zeus V, Kwasnitschka L, et al. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect Genet Evol. 2016;37:108–116.
- Reusken CB, Lina PH, Pielaat A, et al. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010;10(8):785–791.
- Kivistö I, Tidenberg EM, Lilley T, et al. First report of coronaviruses in Northern European bats. Vector Borne Zoonotic Dis. 2020;20(2):155–158.
- Lazov CM, Chriél M, Baagøe HJ, et al. Detection and characterization of distinct alphacoronaviruses in five different bat species in Denmark. Viruses. 2018;10(9):486.
- Lazov CM, Belsham GJ, Bøtner A, et al. Full-genome sequences of alphacoronaviruses and astroviruses from myotis and pipistrelle bats in Denmark. Viruses. 2021;13(6):1073.
- Kemenesi G, Dallos B, Görföl T, et al. Molecular survey of RNA viruses in Hungarian bats: discovering novel astroviruses, coronaviruses, and caliciviruses. Vector Borne Zoonotic Dis. 2014;14(12):846–855.
- Drexler JF, Gloza-Rausch F, Glende J, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84(21):11336–11349.
- Pauly M, Pir JB, Loesch C, et al. Novel alphacoronaviruses and paramyxoviruses cocirculate with type 1 and Severe Acute Respiratory System (SARS)-related betacoronaviruses in synanthropic bats of Luxembourg. Appl Environ Microbiol. 2017;83(18):e01326–17.
- Hardmeier I, Aeberhard N, Qi W, et al. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PloS One. 2021;16(6):e0252534.
- Falcón A, Vázquez-Morón S, Casas I, et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156(10):1883–1890.
- Wong A, Li X, Lau S, et al. Global epidemiology of bat coronaviruses. Viruses. 2019;11(2):174.
- Finnemore M, Richardson PW. Catching bats. In: Mitchell-Jones AJ, McLeish AP, editors. The bat workers’ manual, joint nature conservation committee. Peterborough: UK; 1999. p. 33–38.
- Dietz C, Kiefer A. Bat of Britain and Europe. Vol. 400. London: Bloomsbury Publishing; 2016.
- Escutenaire S, Mohamed N, Isaksson M, et al. SYBR Green real-time reverse transcription-polymerase chain reaction assay for the generic detection of coronaviruses. Arch Virol. 2007;152(1):41–58.
- Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition transversion and G+C-content biases. Mol Biol Evol. 1992;9(4):678–687.
- Boyer F, Mercier C, Bonin A, et al. obitools: a unix -inspired software package for DNA metabarcoding. Mol Ecol Resour. 2016;16(1):176–182.
- Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math Life Sci. 1986;17:57–86.
- Kunz TH, Braun de Torrez E, Bauer D, et al. Ecosystem services provided by bats. Ann N Y Acad Sci. 2011;1223(1):1–38.
- Maine JJ, Boyles JG. Bats initiate vital agroecological interactions in corn. Proc Natl Acad Sci. 2015;112(40):12438–12443.
- de la Fuente J, de Mera IGF, Gortázar C. Challenges at the host-arthropod-coronavirus interface and COVID-19: a One Health approach. Front Biosci. 2021;26(8):379–386.
- Banerjee A, Baker ML, Kulcsar K, et al. Novel insights into immune systems of bats. Front Immunol. 2020;11:26.
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying. 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–544.
- Morens DM, Taubenberger JK, Fauci AS. Universal Coronavirus Vaccines-An Urgent Need. N Engl J Med. 2021;386(4):297–299.
- Middleton DJ, Morrissy CJ, Van Der Heide BM, et al. Experimental Nipah virus infection in pteropid bats (Pteropus poliocephalus). J Comp Pathol. 2007;136(4):266–272.
- Halpin K, Hyatt AD, Fogarty R, et al. Henipavirus Ecology Research Group. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am J Trop Med Hyg. 2011;85(5):946.
- Munster VJ, Adney DR, Van Doremalen N, et al. Replication and shedding of MERS-CoV in Jamaican fruit bats (Artibeus jamaicensis). Sci Rep. 2016;6(1):21878.
- Paweska JT, Storm N, Grobbelaar AA, et al. Experimental inoculation of Egyptian fruit bats (Rousettus aegyptiacus) with Ebola virus. Viruses. 2016;8(2):29.
- Schuh AJ, Amman BR, Sealy TK, et al. Egyptian rousette bats maintain long-term protective immunity against Marburg virus infection despite diminished antibody levels. Sci Rep. 2017;7(1):8763.
- Healy K, Guillerme T, Finlay S, et al. Ecology and mode-of-life explain lifespan variation in birds and mammals. Proc Royal Soc B. 2014;281(1784):20140298
- Gorbunova V, Seluanov A, Kennedy BK. The world goes bats: living longer and tolerating viruses. Cell Metab. 2020;32(1):31–43.
- Kacprzyk J, Hughes GM, Palsson-McDermott EM, et al. A potent anti-inflammatory response in bat macrophages may be linked to extended longevity and viral tolerance. Acta Chiropt. 2017;19(2):219–228.
- Haake C, Cook S, Pusterla N, et al. Coronavirus infections in companion animals: virology, epidemiology, clinical and pathologic features. Viruses. 2020;12(9):1023.
- Menachery VD, Yount BL, Debbink K, et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med. 2015;21(12):1508–1513.
- Gortazar C, Reperant LA, Kuiken T, et al. Crossing the interspecies barrier: opening the door to zoonotic pathogens. PLoS Pathog. 2014;10(6):e1004129.
- Wolfe ND, Daszak P, Kilpatrick AM, et al. Bushmeat hunting, deforestation, and prediction of zoonotic disease. Emerg Infect Dis. 2005;11(12):1822–1827.