
ABSTRACT
Breast cancer is one of the extensively diagnosed cancers and the dominating reason of women’s cancer death globally. Selective estrogen receptor modulators (SERMs) such as Tamoxifen approved by the Food and Drug Administration (FDA) for the therapy of+ breast tumors may cause negative side effects . Hence, there is urgent demand for more effective drugs to combat this disease. Molecular docking screening in combination with ADMET and drug likeness properties were utilized to examine the potency of 57 di-aryl and 2-anilino pyridinamine analogs as ER+ protein receptor inhibitors. Nineteen compounds were found to have better docking scores than the control drug (Tamoxifen, MolDock score = −145.933, Rerank score = −112.169) among which compound 31 emerged as the most effective and potent candidate with best docking scores (MolDock score = −170.206 and Rerank score = −139.238). Furthermore, molecular dynamic simulation studies was performed using Desmond 2019–4 software package to justify the docking study using the docked complex of compound 31, the results confirm the rigidity of the protein-ligand structure during the simulation. The compounds possess drug-likeness and good ADMET properties as indicated by the results of pharmacokinetics studies performed as such they may be utilized as effective drug candidates for breast cancer treatment..
Introduction
Cancer is one of the most problematic and crucial health maladies that affect the globe. The undisputed concern about cancer and mortality rate is significantly expanding [Citation1]. Recently, the targeted treatment of cancers such as breast cancer has been authorized. These inhibitive remedies neutralize cancer cells’ expansion by altering the specific molecules responsible for tumor growth and advancements, and as such, they curtail the damaging impacts of other nonselective chemotherapies and withstand the resistance reproduced by the prevailing anticancer medications [Citation2]. Cancer remediation comprises numerous procedures that include prevention of angiogenesis that has been established to be a desirable scenario for inhibiting the progression of the tumor [Citation3]. Breast cancer is one of the extensively identified cancers and the dominating reason for women’s cancer death globally according to GLOBOCAN 2012 [Citation1]. One of the substantial superfamilies of nuclear receptors called estrogen receptor-α (ERα) is responsible for at least 70% of breast malignancy cases, and these categories of victims are classified as ER-positive (ER+) [Citation4]. The continual activation of ERα by estrogens influences the multiplication of tumor cells, and hence, the blocking up of ER signaling by competitively binding to ER with anti-estrogens or estrogen deprivation is an important therapeutic method [Citation5].
Selective estrogen receptor modulators (SERMs), which can attach with the ER in breast tissue such as Tamoxifen and Raloxifene, are employed for the extermination of ER+ breast tumors [Citation6], but they may cause negative side effects due to their estrogenic properties on some other normal cells [Citation7]. Furthermore, the fact that the majority of the ER+ breast cancer sufferers suppress these drugs during the initial duration of remedy contributes to a substantial complication toward ER+ breast cancer therapy. Consequently, there is a remarkable demand for the evolution of modern drugs to enhance the medicinal impact on breast cancer inhibition [Citation8].
Nitrogen-containing heterocyclics have attracted attention due to their advanced biological activities. This is due to the fact that the electron-rich nitrogen heterocycle is able to receive and contribute proton readily as well as easily form various weak interactions such as hydrogen bond, dipole-dipole interactions, hydrophobic effects, van der Waals forces and Pi-stacking interactions. Ability to form these interactions has improved their significance in the medicinal chemistry field and allows them to bind with a diversity of enzymes and receptors in biological targets with high affinity due to their enhanced solubility [Citation9]. Approved anti-cancer drugs such as Imatinib, Bafetinib, Nilotinib, Pazopanib, Mocetinostat and Brigatinib contain 2-pyridinamine scaffold in their parent structures [Citation10].
Over the past few decades, new drugs have been discovered through random screening and experimental observances on the impacts of natural products on known illnesses; although the method is inadequate, it paved the way for the designation of many essentials drugs. Overall, this process takes time, is laborious and costly, and the average cost of developing new drugs ranges from between 1 and 2 billion USD and can take 10–17 years [Citation11] because it has to pass through all phases for the development of new drug, from target discovery to drug registration. The likelihood of a drug candidate gaining access to the market after entering the first stage of clinical trials decreased from 10% in the 2002–2004 period to about 5% between 2006 and 2008, representing a 50% decrease in only 4 years [Citation12]. Thus, researchers are frequently capitalizing on the advancements of recent techniques to boost the efficiency of the drug discovery procedure [Citation13].
The computer-aided drug design (CADD) process, which utilizes molecular modeling processes, was employed to improve the effectiveness in the designation of new drugs because it employs in silico simulations. Molecular modeling enables the examination of several molecules in a lesser time, clarifying their modes of interaction with targets of pharmacological interest prior to their synthesis. The procedure authorizes the simulation and projection of numerous fundamental factors, such as toxicity, activity, bioavailability and efficacy, prior to conducting in vitro testing on the compounds, thus enabling better planning and direction of the experimenters [Citation14].
Molecular docking analysis reveals how two or more molecular structures interact with each other, for instance, how drug and enzyme or receptor of protein fit together. Molecular docking software packages are purposely utilized in drug design studies. The most vital application of docking software is virtual screening, which chooses favorable molecules from a particular database for advanced exploration. This place mandates the use of the computational approach, which must be quick and credible [Citation15].
This research work is aimed at performing a virtual screening on some series of di-aryl and 2-anilino pyridinamine derivatives as potent inhibitors of ER+ receptor in breast cancers through molecular docking simulation studies and studying their pharmacokinetics and ADMET properties.
Materials and methods
Software packages and online tools utilized in this research
The entire research was carried out using a HP laptop equipped with a dual-core Intel (R) PENTIUM (R) B940 CPU processor running at 2.0 GHz and 4.0 GB of RAM running on Windows 8. The following software packages were used in this research: Chemdraw 19.1, SPARTAN “14 v 1.1.0, Molegro Virtual Docker (MVD) and Discovery Studio. SwissADME and pkCSM online web tools were utilized for evaluating the pharmacokinetics and ADMET properties of the compounds.
Selection of molecules and sketching of their 2-dimensional structures
A series of fifty seven diaryl and anilino pyridinamine derivatives were obtained from Refs. Citation10 and Citation16. All the compounds were tested against the MCF-7 breast cancer cell line, which contains a vast number of estrogen receptors (ER+] and found to be biologically active. Cambridge University-Developed ChemDraw software was employed for the construction of 2- dimensional structures of all the compounds. The structural formulae of the reported compounds are given in Supplementary Table S1.
Transformation of 2D to 3D and geometry optimization
The sketched 2D structures of all the molecules were imported to the Spartan 14.0 software interface, which enables the transformation of their structures from 2D to 3D format. Geometry optimization of the compounds was carried out by employing Density functional theory Quantum mechanical calculations utilizing the B3LYP/631 G* basis set. Geometry optimization was performed in order to find the most stable conformers of all the molecules. The results were saved in a separate folder in PDB format.
Protein download and preparation
The crystal structure of the protein cocrystallized with Tamoxifen (reference drug) was downloaded from the protein data bank (PDB ID = 3ERT) (https://www.rcsb.org/) [Citation17].The protein was imported into the Molegro Virtual Docker (MVD) workspace for preparation where the amino acid residues with structural error were repaired/rebuilt. The cocrystallized ligand and water molecules were eliminated.
Molecular docking process
Molegro Virtual Docker software program produces more accurate and reliable results compared to other docking software packages as such it is employed for the ligand-protein docking simulation in this research work. Prior to the docking process, the prepared protein was imported to the MVD workspace, surface was created and cavities were detected. The binding site of the 3ERT receptor was predicted and was set inside a restricted sphere of volume 137.216 and surface 308.48 with a radius of 15 Å. The ligands preparation was performed from the default by setting if missing to; assign bonds, assign bond orders and hybridization, assign charges and assign tripos atom types as well as creating explicit Hydrogens and detecting flexible torsions in the ligands. The prepared ligands as well as the reference drug (Tamoxifen) were imported onto the MVD surface, and docking was carried out by utilizing a grid resolution of 0.30 Å. The Root Mean Square Deviation (RMSD) threshold was set as 2.00 Å for multiple cluster poses with 100.00 energy penalty values. The docking algorithm was set for a maximum of 1500 iterations with a maximum population size of 50. The docking simulation was run for a minimum of 50 times for the 10 poses, and the best poses were determined based on the set scoring functions such as the MolDock score, rerank score and E-H-bond [Citation18]. Visualization of various intermolecular interactions such as hydrogen bond, hydrophobic, alkyl, Pi-alkyl, halo bonds and aryl interactions was performed by utilizing Discovery studio version 3.5 software program.
ADMET and drug-likeness properties
Accessible online web servers such as SwissADME (http://www.swissadme.ch/index.php) and pkCSM (http:// structure. bioc.cam. ac. uk/ pkcsm) are utilized for the evaluation of the drug-likeness and ADMET properties of the compounds under study. The websites enable researchers to detect the novel drug candidate, to lessen the number of empirical experimentations and to upgrade the success rate [Citation19,Citation20]. In this study, Lipinski’s rule of five (ROF) is utilized as the principal screening step for the drug-likeness properties followed by computing the central ADMET properties, which are measures of the pharmacokinetics of the molecules under study.
Results and discussion
Molecular docking simulation studies were performed between fifty seven (57) series of diaryl and 2-anilino pyridinamine derivatives and the binding pocket of ER+ protein receptor (pdb id = 3ERT). Molegro Virtual docker (MVD) software was employed for this task due to its ability to produce better and more accurate results compared to other docking software packages. Tamoxifen (control drug) was also redocked onto the active site of the 3ERT receptor to justify the docking protocol. Nineteen (19) compounds were identified as potential hit candidates because they have higher MolDock and Rerank Scores than Tamoxifen (MolDock score = −145.933 and Rerank score = −112.169). The MolDock and Rerank scores and energy of the hydrogen bond interactions of all the compounds are presented in , and the results of interactions of the potential hit compounds and 3ERT receptor are shown in . The 3D structures of the prepared 3ERT receptor and compound 1 are shown in , respectively.
Figure 1. 3D structure of the 3ERT receptor.
Figure 2. 3D structure of compound 1.
Figure 3. 3D and 2D representations of Compound 3 in the active site of the 3ERT receptor.
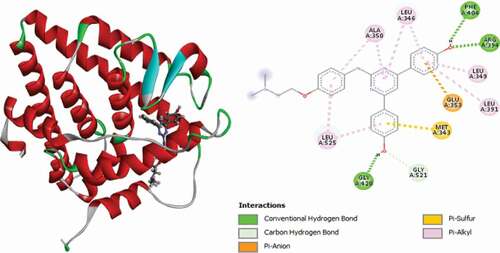
Table 1. Docking scores of the analyzed compounds against the 3ERT receptor with Tamoxifen as the reference
Table 2. Various interactions between the potential hit compounds and the active site of the 3ERT receptor
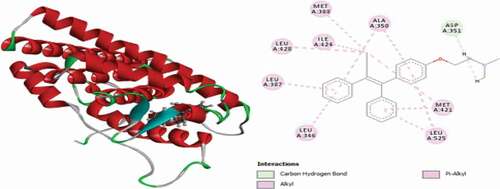
Compound 31 has the best docking scores (MolDock score = −170.206 and Rerank score = −139.238). It was observed to link with the receptor through three (3) conventional hydrogen bonds with LEU536 at a distance of 2.64 Å, GLY420 at a distance of 1.50 Å and GLU353 Å at a distance of 2.03 Å. Two (2) carbon-hydrogen bonds are also detected with VAL534 at a distance of 2.23 Å and VAL533 residue at a distance of 2.22 Å, respectively. The compounds also formed Pi-Sulfur electrostatic interaction with MET343 residue at 5.24 Å. Two (2) Alkyl interactions are also found with LEU536 and VAL533 amino acid residues, and Pi-alkyl interactions with LEU525, LEU346, ALA350, LEU349 and LEU387. 3D and 2D representations of compound 31 are shown in .
Compound 18 is the second best ligand (MolDock score = −168.434 and Rerank score = −122.842). It was found to interact with the receptor via two (2) conventional hydrogen bonds with GLU353 and GLY420 at distances of 2.37 Å and 1.51 Å, respectively. ASP351 forms two (2) carbon-hydrogen bonds at distances of 2.98 Å and 2.82 Å, and other carbon-hydrogen bonds are with GLY521 and ALA350 at distances of 2.11 Å and 2.21 Å. It forms Pi-Alkyl interactions with LEU346, LEU349, ALA350, LEU387 and LEU525. 3D and 2D representations of Compound 18 in the active site of the 3ERT receptor are depicted in .
The 3D and 2D representations of compound 24 (Moldock score = −164.822 and Rerank score = −135.762) in the binding pocket of the 3ERT receptor are shown in . It is observed to interact with the 3ERT protein receptor via two (2) conventional hydrogen bonds with GLY420 at a distance of 1.67 Å and GLU353 at a distance of 2.14 Å, one carbon-hydrogen bond with GLY521 at a distance of 2.23 Å, one Pi-anion electrostatic interaction with ASP351 at a distance of 4.55 Å and one (1) Pi-sulfur interaction with MET343 at a distance of 5.14 Å. LEU536 and LEU539 form two (2) alkyl interactions, and finally, LEU525, LEU346, ALA350, LEU349 and LEU387 form Pi-alkyl interactions with the compound.
Compound 9 has promising docking scores (Moldock score = −162.781 and Rerank score = −135.322). It is found to interact with the receptor through two (2) conventional Hydrogen bonds with GLU353 at a distance of 2.26 Å and GLY420 at a distance of 1.66 Å and one (1) carbon-hydrogen bond with GLY521 at a distance of 2.28 Å. It forms a Pi-Sulfur interaction with MET343 at a bond distance of 5.11 Å, and other interactions observed include three (3) alkyl interactions with MET522, LEU525 and LYS529 and five (5) Pi-alkyl interactions with LEU346, LEU349, ALA350, LEU387 and LEU525. 3D and 2D representations of compound 9 in complex with the 3ERT protein receptor is depicted in .
Compound 27 (MolDock score = −161.015 and Rerank score = −132.486) is bound to the receptor via two (2) conventional hydrogen bonds, with GLU353 at a distance of 2.18 Å and with GLY420 at a distance of 1.51 Å. It also formed a carbon-hydrogen bond with GLY521 at a distance of 2.25 Å, one Pi–Sulfur interaction with MET343 at a bond distance of 5.21 Å and one Pi-Pi stacked interaction with TRP383 at a distance of 5.31 Å. Other hydrophobic interactions observed are alkyl linkages with VAL533 and LEU536 and Pi-alkyl interactions with LEU346, LEU349, ALA350, LEU387 and LEU525 amino acid residues. 3D and 2D representations of compound 27 in the active site of the 3ERT protein receptor are shown in .
Compound 8 (MolDock score = −159.641 and Rerank score = −129.307) is bound to the receptor through two (2) conventional hydrogen bonds, with GLY420 at a distance of 1.75 Å and PHE404 at 2.04 Å, a single carbon-hydrogen bond with LEU525 at a distance of 3.05 Å, two (2) electrostatic Pi-anions with ASP351 at a distance of 4.83 Å and GLU353 at a distance of 4.31 Å and two (2) Pi-Sulfur interactions with MET343 at distances of 5.26 and 5.90 Å. Other hydrophobic interactions observed include two (2) Alkyl interactions with LEU525 and LYS529 and Pi-Alkyl interactions with LEU525, LEU346, ALA350, LEU349 and LEU391 amino acid residues. shows the 3D and 2D representations of Compound 8 in the active site of the 3ERT receptor.
shows the representation of Compound 23 (Moldock score = −156.108 and Rerank score = −119.832) in the active site of the 3ERT receptor. It is observed to link with the receptor via a single conventional hydrogen bond with GLU353 at a bond distance of 2.31 Å, GLY420 amino acid residue forms double carbon-hydrogen bonds at bond distances of 2.89 Å and 2.24, also LYS520 forms a pair of carbon-hydrogen bonds at distances of 3.05 Å and 2.56 Å, respectively, and it also forms Pi-Sulfur interactions with MET343. Other hydrophobic interactions observed include one Pi-Pi stacked interaction with TRP383, two (2) alkyl interactions with MET522 and LEU525 and Pi-alkyl interactions with LEU349, LEU387, LEU346, ALA350 and LEU525.
Compound 12 (MolDock score = −152.327 and Rerank score = −125.731) interacted with the receptor through three (3) conventional hydrogen bonds, ARG394 at a distance of 1.87 Å, PHE404 at a distance of 2.39 Å and GLY420 at a distance of 1.70 Å and two (2) carbon-hydrogen bonds with GLY521 at a distance of 2.35 Å and GLU380 at a distance of 2.88 Å. MET343 forms the Pi-Sulfur interaction type at 5.17 Å. Other hydrophobic interactions observed are one (1) Pi-Pi stacked with TRP383, single alkyl interaction with LEU536 and Pi-alkyl interactions with TRP383, LEU346, LEU349, ALA350, LEU387 and LEU525 amino acid residues. 3D and 2D representations of compound 12 in the binding pocket of the 3ERT receptor are given in .
Compound 21 (MolDock score = −150.714 Rerank score = −124.160) interacts with the receptor via three (3) conventional hydrogen bonds, PHE404 at a distance of 2.37 Å, GLY420 at a distance of 1.90 Å and LEU346 at a bond distance of 2.99 Å, single carbon-hydrogen bond with GLY521 at a distance of 2.34 Å and single Pi-Sulfur interaction with MET343 at a distance of 5.01 Å. Other hydrophobic interactions observed include LYS529, which forms two (2) alkyl interactions, VAL533, which forms another alkyl interaction, and TYR526, LEU349, ALA350, LEU387, LEU346 and LEU525, which form Pi-alkyl interactions. 3D and 2D representations of Compound 21 in the active site of the 3ERT receptor are shown in .
Compound 3 (MolDock score = −150.523 Rerank score = −119.071) is observed to interact with the receptor via three (3) conventional hydrogen bonds, with ARG394 at a distance of 1.97 Å, PHE404 at a distance of 2.26 Å and GLY420 at a distance of 1.65 Å, a single carbon-hydrogen bond with GLY521 at a distance of 2.29 Å, single electrostatic Pi-anion interaction with GLU353 at a distance of 4.34 Å and single Pi-Sulfur interaction with MET343 at a distance of 5.09 Å. Other hydrophobic Pi-alkyl interactions are also observed with LEU346, LEU349, LEU391, ALA350 and LEU525 amino acid residues. shows the 3D and 2D representations of Compound 3 in the active site of the 3ERT receptor.
Compound 5 (Moldock score = −149.681 and Rerank score = −121.819) linked with the receptor via two (2) conventional hydrogen bonds with GLY420 at a distance of 1.99 Å and PHE404 at a distance of 2.20 Å, four carbon-hydrogen bonds with GLY521 at a distance of 2.52 Å, LEU525 forms two (2) interactions at distances of 2.60 Å and 1.97 Å and MET528 at a distance of 2.78 Å, single electrostatic Pi-anion interaction with GLU353 at a distance of 4.31 Å and double Pi-Sulfur interactions with MET343. Other hydrophobic interactions observed include three alkyl interactions with CYS530, VAL533 and LYS529 and several Pi-alkyl interactions with LEU346, ALA350, LEU349, LEU391 and LEU525 amino acid residues. 3D and 2D representations of Compound 5 in the active site of the 3ERT receptor are shown in .
Compound 15 (Moldock score = −148.410 Rerank score = −115.575) interacted with the receptor via two (2) conventional hydrogen bonds with GLU353 at a distance of 2.25 Å and GLY420 at a distance of 1.74 Å, single carbon-hydrogen bond with GLY521 at a distance 2.29 Å and single Pi-Sulfur interactions with MET343 at distance of 5.09 Å. Three alkyl interactions are also observed with VAL533, LEU536 and LEU539 and several Pi-Alkyl interactions with LEU346, LEU349, ALA350, LEU387 and LEU525 residues. depicts the 3D and 3D representation of compound 15 in the active site of the 3ERT receptor.
Compound 7 (MolDock score = −145.717 and Rerank score = −117.982) interacted with the receptor through single conventional hydrogen bond with VAL534 at a distance of 1.85 Å, two (2) carbon-hydrogen bonds with LEU346 at a distance of 2.43 Å and GLU353 at a distance of 2.62 Å and a Pi-Sulfur interaction with MET343 at a distance of 5.42 Å. Other hydrophobic interactions observed include a Pi-Pi T-shaped interaction with TRP383, amide Pi-stacked with PRO353 and LEU536, Alkyl interactions with ALA350, LEU387 and LEU391 and Pi-alkyl interactions with VAL533, LEU536, LEU525, MET522 and ALA350. 3D and 2D representations of Compound 7 in the active site of the 3ERT receptor are shown in .
The interaction pattern of compound 29 (MolDock score = −145.925 and Rerank score = −114.010) with the receptor is through a single conventional hydrogen bond with LEU525 at a distance of 1.99 Å, two (2) carbon-hydrogen bonds with PRO353 at a distance of 3.07 Å and MET522 at a distance of 2.95 Å, single electrostatic Pi-cation interaction with LYS529 at a distance of 4.93 Å, single Pi-donor hydrogen bond with CYS530 at a 2.63 Å , two Pi-Sulfur interactions with MET522 and CYS530 and Pi-alkyl hydrophobic interactions with TYR526, LYS529, CYS530, VAL533, LYS529, VAL533 and PRO535 residues. shows the 3D and 2D representation of compound 29 in the active site of the 3ERT receptor.
Compound 50 (MolDock score = −147.708 and Rerank score = −116.954) interacted with the binding site of the ER+ receptor via single conventional hydrogen bonds, two (2) carbon-hydrogen bonds, single electrostatic Pi-Anion and Pi-Sulfur and several hydrophobic Pi-Alkyl interactions. Carbonyl oxygen of the pyrazolidinone group forms a conventional hydrogen bond with HIS524 residue at a distance of 1.99 Å and forms two (2) carbon-hydrogen bonds with GLY420 and GLY521 at a distance of 2.33 Å. The phenyl rings intercalated in space and forms an electrostatic Pi-Anion interaction with GLU353 residue at a distance of 4.2898 Å and Pi-Sulfur interaction with MET343 at a distance of 5.74 Å. LEU346, LEU387, LEU391, ALA350 and LEU525 amino acid residues form hydrophobic Pi-Alkyl interactions. 3D and 2D representations of compound 50 in the binding site of the ER+ receptor kinase is shown in .
Compound 51 (MolDock score = −145.683 and Rerank score = −116.258) interacted with the active site of the receptor through a single conventional hydrogen bond, Pi-Anion and Pi-Sulfur, and hydrophobic Alkyl and Pi-Alkyl interactions. The nitrogen atom of the aniline group forms a conventional hydrogen bond with LEU346 at a distance of 2.32 Å. The pyrazole group intercalated in space and forms an electrostatic Pi-Anion interaction with GLU353 at a distance of 4.62 Å, and aniline benzene ring intercalated in space and forms a Pi-Sulfur interaction with MET343 residue at a distance of 5.87 Å. LEU384, LEU387, MET388, LEU346, LEU349 and LEU391 residues formed Alkyl interactions, while PHE404, MET343, MET421 and LEU525 residues formed hydrophobic Pi-alkyl interactions. 3D and 2D representations of compound 51 in the active site of the ER+ receptor are shown in .
Compound 52 (MolDock score = −148.662 Rerank score = −116.942) is observed to have interacted with the ER+ binding pocket through single conventional hydrogen bond, carbon-hydrogen bond, electrostatic Pi-Anion and Pi-Sulfur interactions and numerous Pi-Alkyl hydrophobic interactions. Carbonyl oxygen of the pyrazolinone group forms conventional and carbon-hydrogen bonds with HIS524 and GLY420 residues at distances of 2.01 Å and 2.30 Å, and phenyl rings attached to the pyrimidine group intercalated in space and form an electrostatic Pi-Anion interaction with GLU353 at a distance of 4.09 Å and Pi-Sulfur interaction with MET343 at a distance of 5.79 Å. LEU346, LEU349, LEU387, LEU391, ALA350 and LEU525 amino acid residues formed Pi-Alkyl hydrophobic interactions. depicts the 3D and 2D interactions of compound 52 in the binding pocket of the ER+ receptor.
Compound 54 (MolDockscore = −150.696 and Rerank score = −119.567) is found to have interacted with the binding site of the ER+ receptor through two (2) conventional hydrogen bonds and single Pi-Sulfur and several Pi-Alkyl hydrophobic interactions. ARG394 and LEU346 form two conventional hydrogen bonds with the cyano nitrogen atom of the pyrazolinone group and the aniline nitrogen atom at 2.72 Å and 2.10 Å. The anilino benzene ring intercalated in space and forms a Pi-Sulfur interaction with MET343 at 5.82 Å. MET421, LEU525, LEU346 and ALA350 residues formed hydrophobic Pi-Alkyl interactions. 3D and 2D representations of Compound 54 in the active pocket of the ER+ receptor are presented in .
Compound 55 is bound to the active site of the ER+ receptor through two (2) conventional hydrogen bonds and single Pi-Sulfur and numerous Alkyl and Pi-Alkyl hydrophobic interactions. ARG394 and LEU346 formed two (2) conventional hydrogen bonds with the cyano nitrogen atom of the pyrazolinone group and the anilino hydrogen atom at distances of 2.16 and 2.24 Å. MET343 forms Pi-Sulfur interactions with the aniline phenyl ring at a distance of 5.83 Å. LEU346, LEU349 and LEU391 residues formed Alkyl and PHE404, MET421, LEU525, LEU346 and ALA350 formed Pi-Alkyl hydrophobic interactions. represent the 3D and 2D interactions of compound 55 with the binding site of the ER+ receptor.
For the rationalization of the docking simulation results, the control drug (Tamoxifen), which is also the cocrystallized ligand, was also redocked into the binding pocket of the 3ERT protein receptor. It is found to have a MolDock score of −145.933 and a Rerank score of −112.169, and it is found to interact with the receptor via single carbon-hydrogen bond with ASP351 amino acid residue at a bond distance of 1.86477 Å, hydrophobic alkyl interactions with MET388, MET421, ILE424 and LEU428 and Pi-alkyl interactions with ALA350, LEU525, MET421, LEU346 and LEU387 amino acid residues. 3D and 2D representations of Tamoxifen in the active site of the 3ERT receptor are given in .
The study disclosed that H-bonding is the fundamental driving force that regulates the interactions that exist between the selected target hit compounds and the 3ERT protein receptor and also the docking scores of the compounds vary directly with the number of hydrogen bonds. The conventional and carbon-hydrogen bonds associated with some of the selected analog, the number of amino acids linked was found to be better compared to the control drug (Tamoxifen) as depicted in and consecutively, there are great correspondences. This might explain the better docking scores of the selected compounds compared to Tamoxifen. Hence, the target hit molecules can be utilized as potent inhibitors of ER+, displaying competitive inhibition with Tamoxifen as evident from the result of the docking simulation studies [Citation19].
Figure 4. 3D and 2D representations of Compound 5 in the active site of the 3ERT receptor.
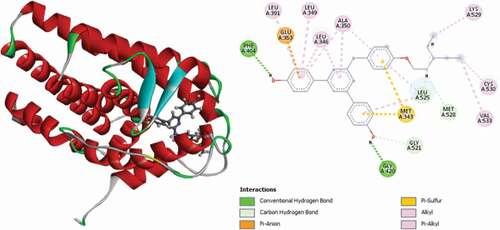
Figure 5. 3D and 2D representations of Compound 7 in the active site of the 3ERT receptor.
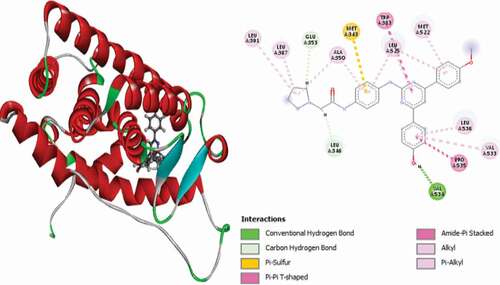
Figure 6. 3D and 2D representations of Compound 8 in the active site of the 3ERT receptor.
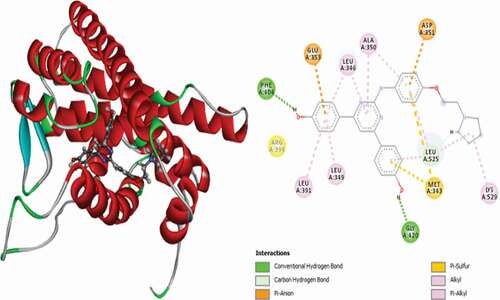
Figure 7. 3D and 2D representations of Compound 9 in the active site of the 3ERT receptor.
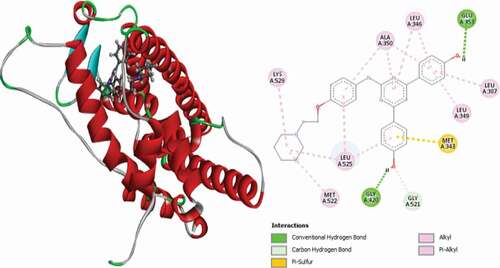
Figure 8. 3D and 2D representations of Compound 12 in the active site of the 3ERT receptor.
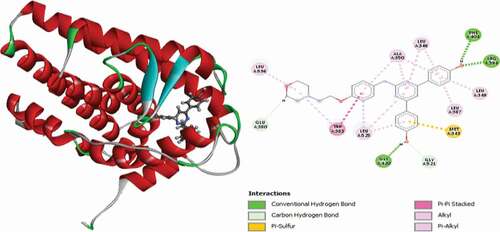
Figure 9. 3D and 2D representations of Compound 15 in the active site of the 3ERT receptor.
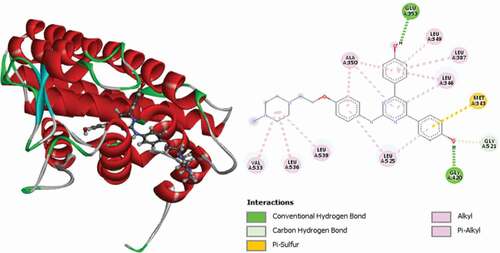
Figure 10. 3D and 2D representations of Compound 18 in the active site of the 3ERT receptor.
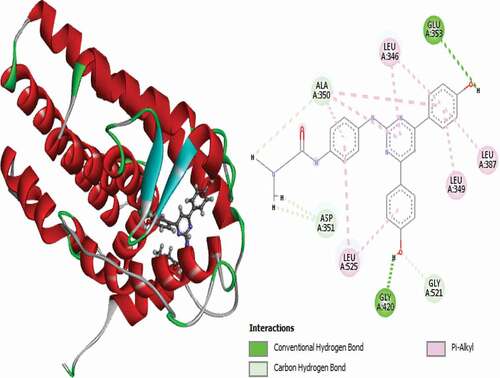
Figure 11. 3D and 2D representations of Compound 21 in the active site of the 3ERT receptor.
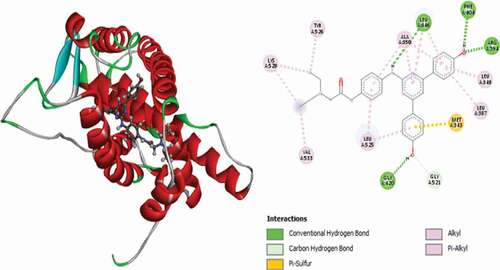
Figure 12. 3D and 2D representations of Compound 23 in the active site of the 3ERT receptor.
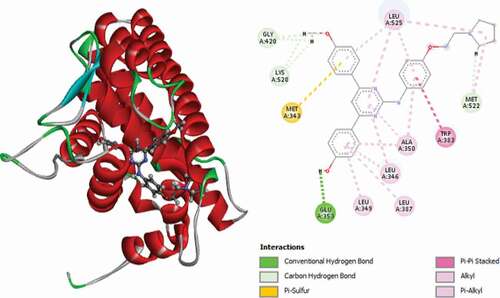
Figure 13. 3D and 2D representations of Compound 24 in the active site of the 3ERT receptor.
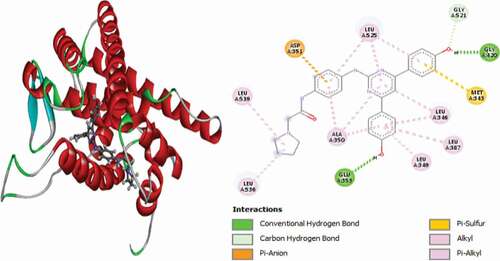
Figure 14. 3D and 2D representations of Compound 27 in the active site of the 3ERT receptor.
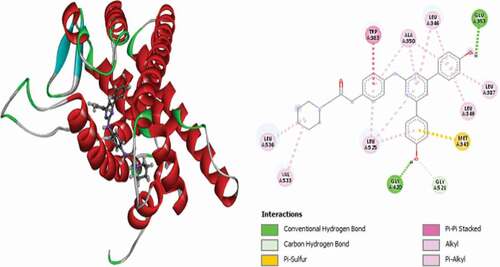
Figure 15. 3D and 2D representations of Compound 29 in the active site of the 3ERT receptor.
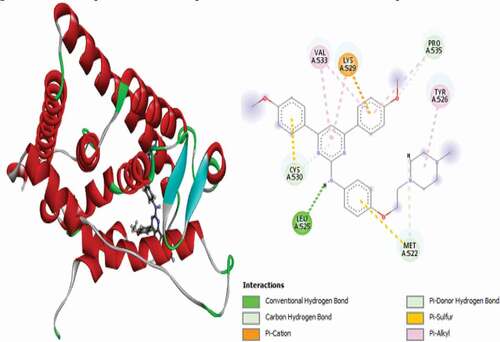
Figure 16. 3D and 2D representations of Compound 31 in the active site of the 3ERT receptor.
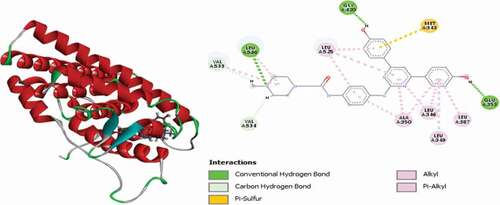
Figure 17. 3D and 2D representations of compound 50 in the binding site of the ER+ receptor.
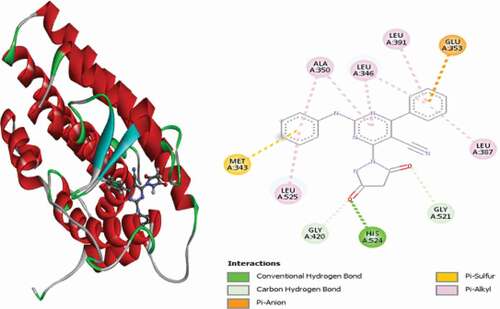
Figure 18. 3D and 2D representations of compound 51 in the active site of the ER+ receptor.
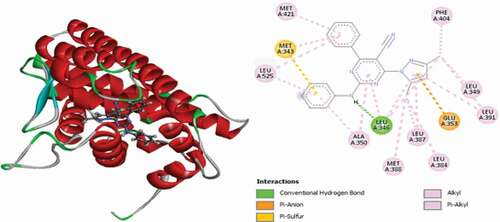
Figure 19. 3D and 2D interactions of compound 52 in the binding pocket of the ER+ receptor.
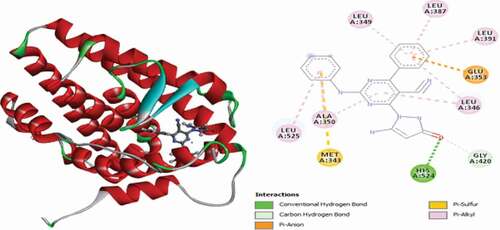
Figure 20. 3D and 2D representations of Compound 54 in the active pocket of the ER+ receptor.
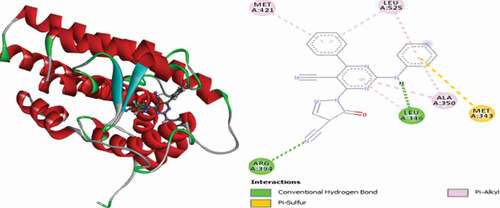
Figure 21. 3D and 2D interactions of compound 55 with the binding site of the ER+ receptor.
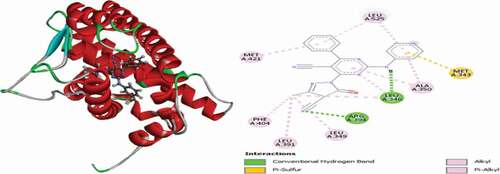
Figure 22. 3D and 2D representations of Tamoxifen in the active site of the 3ERT receptor.
Molecular dynamic (MD) simulation
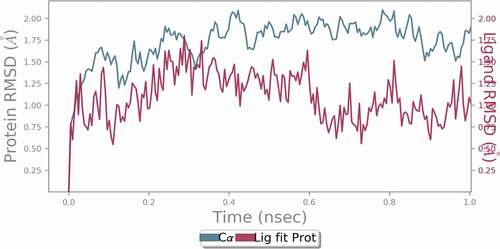
The docked complex of compound 31 was utilized as the initial structure for the molecular dynamic simulation study using Desmond 2019–4 software package in the present research work because it exhibits best docking scores as well as hydrogen bond energy. MD simulation generates a model for the ligand-receptor state very close to the natural environment, which offers a noble opportunity to examine the strength and continuity of the H-bonds profile. Protein and ligand flexibility was taken into consideration in MD simulation, this paves the way for advanced examination of the consistency of the docking outcomes and residues in the binding site of a receptor and their nature of interactions with inhibitors can also be explored during the simulation process [Citation21]. The complex was tested for root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg) and number of H-bonds formed between the inhibitors and the receptor after the simulation process was completed. The system is well equilibrated and remained stable throughout the simulation as revealed by the analysis of trajectory. RMSD values of the backbone of the protein alone and that of the ligand-protein complex were computed to explore the stability of the complex. Final RMSD values of the complex and protein lie below 2.00 Å for the 1 ns simulation, which illustrates the rigidity of the protein structure in the presence of the ligand during MD simulation. The ligand and the protein systems reached equilibrium at 0.28 ns and 0.4 ns, respectively. The RMSD value of the ligand is lower than that of the protein, which illustrates the stability of the ligand in the active site of the protein. shows the RMSD values of the protein and the ligand.
Figure 23. RMSD values of the protein and ligand during 1 ns MD simulation.
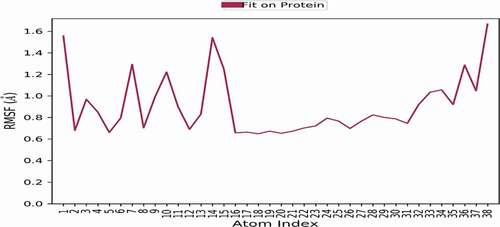
RMSF values of the backbone protein alone and the ligand-protein complex were computed to explore the flexibility of the ER+ backbone. The high value of RMSF suggests more flexibility, while low values illustrate more stability during simulation. show the RMSF values of the protein and ligand-protein complex during 1 ns simulation. It is observed that RMSF values of the complex are lower than those of the protein alone, which confirms the stability of the ligand in the active site of the receptor.
Figure 24. RMSF values of the backbone protein.
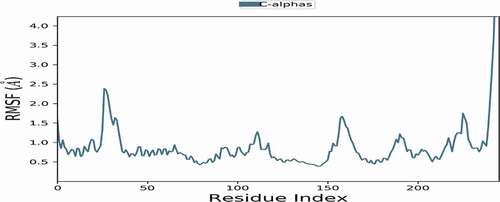
Figure 25. RMSF values of the ligand-protein complex.
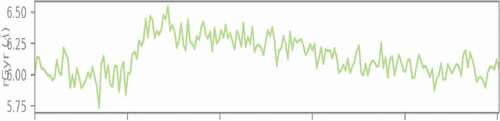
A measure of the ‘extendedness’ of a ligand is termed as its radius of gyration and is equivalent to its principal moment of inertia. Moreover, the radius of gyration of the ligand protein was computed during the course of the simulation and was found to maintain a relatively steady value throughout the simulation, which illustrates the stability of the ligand-protein complex ().
Figure 26. Radius of gyration of the ligand-protein complex during 1 ns dynamic simulation.
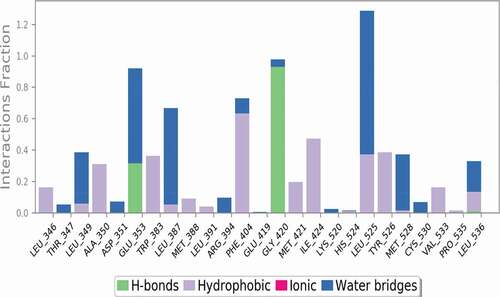
Hydrogen bonds play a significant role in stabilizing the protein-ligand complexes. As the ligands form more hydrogen bonds with the protein, the protein-ligand complex becomes more stable. Hydrogen bond analysis was performed to reveal the stability of the ligand-protein complex and confirms the formation of three (3) H bonds with GLU353, GLY420 and LEU536 amino acid residues, which confirms the stability of the complex as well as justifies the docking result.
Figure 27. Ligand-protein contacts during 1 ns simulation studies.
ADMET and pharmacokinetics studies
To certify that the target hit compounds are the feasible drugs, the ADMET and pharmacokinetics properties were assessed with Tamoxifen as the control drug. An online web tool SwissADME [Citation22] was utilized to compute the drug-likeness properties, and the pkCSM online web page was embraced to formulate the ADMET properties of the selected compounds. The key benchmark used in the selection of drug aspirants in the primary phase of the drug discovery procedure is the estimation of their drug-likeness parameters. This is achieved by correlating the physicochemical properties of a given molecule with its biopharmaceutical aspect inside a human body, mostly, its impact on oral bioavailability [Citation23].
The most original and detailed exploration of drug-likeness properties was performed by Lipinski [Citation24], which gave rise to the popular ‘rule of 5’, which suggests that a compound will be well absorbed or permeated only when its molecular weight (mol. wt.) < 500, its number of hydrogen bond donors (HBD) < 5, its number of hydrogen bond acceptors (HBA) < 10 and its partition coefficient octanol/water Log P < 5. A compound is said to satisfy the rule of five if it does not violate more than two (2) of the rules. Results of the drug-likeness properties of the potential hit compounds are presented in . From the results of the pharmacokinetic studies, all the selected compounds are well absorbed, are permeable and possess drug-likeness properties because seventeen (17) compounds do not violate any of the rules, while the remaining two (29 and 31) violate only one (1) of the rule since their molecular weight exceeded 500.
Table 3. Predicted drug-likeness properties of the selected compounds
Further assessment was executed by using bioavailability score (ABS) standards [Citation25], where all the selected compounds have 0.55 as the computed value. This approach was on the basis of the probability value of a molecule to possess an optimal profile of permeability and bioavailability, where 0.55 designates the compliance with the Lipinski rule of five, and the rat bioavailability value is 55%, which is a greater probabilistic value than 10%. Moreover, the selected compounds were also assessed for their synthetic accessibility, based on a scale that ranges from 1 (very simple to synthesize) and 10 (very difficult to synthesize). The synthetic accessibility for all chosen compounds is around 3.13 to 4.0 (), and hence, the compounds can be easily synthesized.
Additionally, all the selected compounds possess excellent human intestinal absorbance values from 86.088% to 99.312% since these values exceed the minimal recommended values of 30%. The approved values of blood-brain barrier (BBB) and the central nervous system (CNS) permeability are given as > 0.3 to < −1 log BB and > −2 to < −3 log PS. Hence, for any compound, log BB less than −1 illustrates unfavorable dispersal of the drug to the brain, while a value of log BB > 0.3 implies that the drug can cross the blood-brain barrier (BBB). With regard to log PS, a value greater than −2 entails that the compound is able to infiltrate the CNS, while a value less than −3 implies that it might be tough for the compound to induce into the CNS [Citation26]. The results presented in suggested that the chosen compounds have shown a high possibility of crossing the barriers. The biotransformation of a drug in a body is shown by the enzymatic metabolism of the drugs, and hence, it is very fundamental to consider the drug’s metabolism. A class of super enzymes cytochrome P450 play a crucial role in drug’s metabolism. CYP families responsible for the metabolism of drug include 1A2, 2C9, 2C19, 2D6 and 3A4, among which the most important is the 3A4 enzyme in which most of the selected compounds were found to be its substrate as well as inhibitor.
Table 4. Predicted ADMET properties of the selected compounds
An indicator, which explains the connection between the elimination rate of drug and its amount in the body, is called total clearance, and the selected compounds displayed higher values of total clearance, which are within the acceptable range of a drug candidate in the body. Additionally, only six (24, 27, 51, 52, 54 and 55) of the compounds possess some level of toxicity. Finally, the selected compounds showed satisfactory physicochemical and pharmacokinetic ADMET properties. Thus, on the basis of these outcomes, it can be acknowledged that these compounds can be embraced as ER+ inhibitors and drugs on breast cancer in the forthcoming years.
Conclusion
Molecular docking simulation studies were performed on fifty seven (57) series of di-aryl and 2-anilino pyridinamine analog as potential inhibitors of estrogen receptor (ER+) using Molegro Virtual Docker software. The compounds were screened based on their MolDock and Rerank scores. Nineteen (19) of the compounds were found to have better MolDock and Rerank scores than the control drug (Tamoxifen) utilized in the research, and hence, they were tagged as potential hit compounds. Molecular dynamic simulation studies were performed using Desmond 2019–4 software package to justify the docking study using the complex formed between compound 31 and the active site of the ER+ receptor, and the results confirm the rigidity of the protein structure in the presence of the ligand during MD simulation as observed from the RMSD values of the backbone of the protein alone and those of the ligand-protein complex. Lower RMSF values of the complex compared to those of the protein confirm the stability of the ligand in the active site of the receptor. Hydrogen bond analysis performed confirms the formation of three (3) H-bonds with GLU353, GLY420 and LEU536 amino acid residues, which confirms the stability of the complex as well as justifies the docking result. Furthermore, the potential hit compounds were then subjected to ADMET and pharmacokinetic profile studies in order to explore their drug-likeness and pharmacokinetic properties. None of the 19 compounds violated Lipinski’s rule of five (ROF), as such they are well absorbed and permeable and possess drug-likeness properties. Their bioavailability score of 0.55 suggested that they are orally bioavailable and their synthetic accessibility value lies within 3.13 to 4.00, which indicates that they can be easily synthesized. Additionally, the result of ADMET studies suggested that the compounds possess excellent human intestinal absorbance values from 86.088% to 99.312%, since the values exceed the minimal recommended values of 30%, they can cross the blood-brain barrier and can penetrate the central nervous system, they are mostly substrate as well as inhibitors of cytochrome P450 enzymes that are accountable for metabolism of drug and they are easily eliminated from the body system. Additionally, only six (24, 27, 51, 52, 54 and 55) of the potential hit compounds possess some level of toxicity. Hence, the selected compounds could be used as potential drugs for the inhibition of ER+ in breast cancer.
Abbreviations
SERM: Selective Estrogen Receptor Modulators; FDA: Food and Drug Administration; ADMET: Absorption Distribution Metabolism Excretion and Toxicity; ER: Estrogen Receptor; CADD: Computer Aided drug design; MCF-7: Michigan Cancer Foundation-7; DFT: Density Functional Theory: MVD: Molegro Virtual Dock
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Canc J Clin. 2015;65(2):87–108.
- Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Phys. 2008;77:311–319.
- Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21(3):505–515.
- Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Semin Cancer Biol. 2001;11(5):339–352.
- Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72(1):7–25.
- Wang T, You Q, Huang FS, et al. Recent advances in selective estrogen receptor modulators for breast cancer. Mini Rev Med Chem. 2009;9(10):1191–1201.
- Van Leeuwen FE, Benraadt J, Coebergh JW, et al. Risk of endometrial cancer after treatment tamoxifen treatment of breast cancer. Lancet. 1994;343:448.
- Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer- An overview and update. Mol Cell Endocrinol. 2015;418:220–234.
- Kerru N, Gummidi L, Maddila S, et al. AReview on recent advances in nitrogen containing molecules and their biological applications. Molecules. 2020;25(8):1909.
- Liu L, Tang Z, Chengze W, et al. Synthesis and biological evaluation of 4,6-diaryl-2-pyrimidinamine derivatives as anti-breast cancer agents. Bioorg Med Chem Lett. 2018;28(6):1138–1142.
- Leelananda SP, Lindert S. Computational methods in drug discovery. Beilstein J Org Chem. 2016;12:2694–2718.
- Arrowsmith J. A decade of change. Nat Rev Drug Discov. 2012;11(1):17–18.
- Hillisch A, Pineda LF, Hilgenfeld R. Utility of homology models in the drug discovery process. Drug Discov Today. 2004;9(15):659–669.
- Ferreira RS, Glaucius O, Andricopulo AD. Integração das técnicas de triagem virtual e triagem biológica automatizada em alta escala: oportunidades e desafios em P&D de fármacos. Quim Nova. 2011;34(10):1770–1778.
- Yang Y, Qin J, Liu H. Molecular dynamics simulation, free energy calculation and structure-based 3D-QSAR studies of B-RAF kinase inhibitors. J Chem Inform Model. 2011;51(3):680–692.
- Omaima MA, Daabeesb HMG, Badawi WA. 2-Anilinopyrimidine derivatives: design, synthesis, in vitro anti-proliferative activity, EGFR and ARO inhibitory activity, cell cycle analysis and molecular docking study. Bioorg Chem. 2020;99:103798.
- Chang J, Ren H, Zhao M, et al. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur J Med Chem. 2017;138:669–688.
- Thomsen R, Christensen MH. MolDock: a new technique for high accuracy molecular docking. J Med Chem. 2006;49(11):3315–3321.
- Bello Umar A, Uzairu A, Adamu Shallangwa G, et al. Computational evaluation of potent 2 (1Himidazol- 2-yl) pyridine derivatives as potential V600E-BRAF inhibitors. Egypt J Med Hum Genet. 2020;21(1):67. https://doi.org/10.1186/s43042-020-00111-2
- Sagiru Hamza A, Uzairu A, Ibrahim, et al., Tukur Ibrahim and Abdullahi Bello Umar. Chemo‑informatics activity prediction, ligand based drug design, Molecular docking and pharmacokinetics studies of some series of 4, 6‑diaryl‑2‑pyrimidinamine derivatives as anti‑cancer agents. Bull Natl Res Cent. 2021;45(1):167.
- Mohameda TK, Batrana RZ, Elseginyb SA, et al. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg Chem. 2019;85:253–273.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Bickerton GR, Paolini GV, Besnard J, et al. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25.
- Martin YC. A bioavailability score. J Med Chem. 2005;48(9):3164–3170.
- Clark DE. In silico prediction of blood–brain barrier permeation. Drug Discov Today. 2003;8(20):927–933.