
ABSTRACT
The anti-apoptotic Bcl-2 family is intrinsically involved in regulating apoptosis. Over expression of these proteins is associated with cancer. Thus, inhibitors of these proteins will enhance the development of anti-apoptotic drugs. Herein, previously reported 103 Ocimum gratissimum derived phytochemicals were screened against five anti-apoptotic BCL-2 proteins (BCL-2, MCL-1, BCL-B BCL-XL and BFL-1) to identify potential inhibitors of multiple anti-apoptotic Bcl-2 proteins, using static and dynamic docking simulations, molecular dynamics (MD) simulations, clustering and Absorption-Distribution-Metabolism-Excretion-Toxicity (ADMET) filtering analysis. Based on the minimal binding energy and a comparative reference inhibitors binding mode analysis, five lead phytochemicals (FLP) (ursolic acid, beta-sitosterol, luteolin, basilimoside and apigenin 7,4’,dimethyl ether) were identified. Ursolic acid, β-sitosterol and luteolin exhibited higher binding tendencies to the BH3 binding groove of multiple targets. Ursolic acid-Bcl-2 and luteolin-BCL-XL, complexes demonstrated structural stability in the simulated MD environment. Also, the FLP demonstrated favorable ADMET properties. Evidences from previously reported antiproliferative activities of ursolic acid, β-sitosterol and luteolin and results from this study suggest that the anti-proliferative activity of O. gratissimum may be as a result of the synergistic activities of, at least, the FLP. They are recommended for further study as natural-inhibitors against cancers defined by over expression of Bcl-2 family protein.
Introduction
The members of the anti-apoptotic B cell lymphoma 2 (BCL- 2) protein family play key regulatory roles in cellular apoptosis. Deregulation of the normal apoptotic process in the cell, driven by the members of the BCL-2 proteins, is a fundamental element in carcinogenesis [Citation1]. The Bcl-2 proteins are reportedly overexpressed in about half of all human cancers which reveals the involvement of the anti-apoptotic members in carcinogenesis (Yip and Reed 2008). The four BH (BH1, BH2, BH3 and BH4) conserved domains, which were identified from nuclear magnetic resonance (NMR) investigation in 2001 [Citation2], are critical for the signaling functions of these proteins. The BH1-BH3 domain forms a hydrophobic groove, while the BH4 stabilizes the entire structure. The BH3 domain which resides superficially on the anti-apoptotic proteins plays a central role as it accommodates pro-apoptotic partners (Bax/Bak) [Citation3]. On account of this structural built up, substances that interact with the critical amino acid residues in this groove serve as promising inhibitors that may interfere with anti-apoptotic BCL-2 proteins; and thereby promote apoptosis in cancer cells [Citation4]. In this direction, navitoclax [Citation5] and ABT-199 [Citation6] and other drugable compounds have been developed through combinatorial technique and are under way through clinical trials for treatment of cancers [Citation7]. Thus, targeting the BCL- 2 proteins, which are known to perform pivotal functions in the regulation of apoptosis, have been explored toward developing anticancer drugs.
The use of food spices and medicinal herbs as complementary medicine predates human history [Citation8]. Notable among these dietary components is Ocimum gratissimum L, a herbaceous plant in the Labiatae family thought to originate from Asia and Africa [Citation9]. This plant is highly cherished for its health promoting potentials and aromatic nature, and popularly known as scent leaf in Nigeria and some other African regions where it is grown in home gardens as an ingredient in foods such as pastas, salads, soups, vinegars and jellie [Citation10]. Various health benefits of this plant have been documented for over two decades. The several pharmacological activities of the plant have been reported by Priyanka et al. [Citation11]. The ethno-pharmacological uses of scent leaf in cancer therapy have been proven from various in vitro and in vivo studies. The proliferation inhibitory activity of aqueous extracts of O. gratissimum against several cell lines particularly prostate adenocarcinoma (PC-3) cells is well documented [Citation12]. Various solvent fractions from this plant have been reported to impede the tumor growth and progression of breast cancer [Citation13]. Aqueous Ocimum gratissimum extract has been reported to induce apoptosis in human hepatocellular carcinoma cells [Citation14]. Thus, this plant is a promising vegetal source for pro-apoptosis compounds and chemopreventive nutraceuticals in cancers [Citation15]. Several bioactive compounds such as: Eugenol, Thymol, β-caryophyllene, γ-muurolene, γ-terpinene, α-bisabolene, β-selinene, 1.8-cineole, geraniol, and p-cymene have been identified from this plant [Citation16]. In search for phytocompounds with inhibitory potential against the anti-apoptotic BCL-2 proteins, 103 bioactive compounds (previously reported as active constituent of O. gratissimum) were compiled from reports where the chemical constituents were identified and/or isolated [Citation16–24] and review articles on the chemical composition [Citation25,Citation26]) were screened in silico for interaction with five anti-apoptotic targets in cancer. This computer-based screening approach is exceptionally valuable for resource optimization and search space minimization. Such approach undertaken may help provide new evidence for anticancer activity of O. gratissimum.
Materials and Methods
Retrieval and Preparation of Proteins
The following anti-apoptotic BCL-2 proteins (APB2P) were accessed from the Protein Databank (http://www.rcsb.org): BCL-2 (4lvt); BCL-XL (3zlr); MCL-1 (5fdr); BFL-1 (5uul) and BCL-A1 (5whi).
The APB2P were selected as the best proteins for the interactive analysis based on their conformation with a co-crystallized ligand and high resolutions, also the Ramachandran plots of the APB2P were analyzed. The water molecules and bound ligands associated with the protein structures were gotten rid of for efficient docking without interference while the missing hydrogen atoms were added. Employing MGL-AutoDockTools (ADT, v1.5.6), the Kollman charges were added as the partial atomic charge [Citation27]. Thereafter, while nonpolar hydrogens were merged, the polar hydrogens were added to the proteins. This procedure was applied to all proteins and then saved in pdbqt format for the docking calculations.
Ligand preparation
The 103 O. gratissimum- derived compounds (OGDPs) were gathered from extensive literature search. These compounds and the reference compounds were downloaded from the PubChem database (www.pubchem.ncbi.nlm.nih.gov) in Structure Data Format (SDF), and converted to mol2 format using Openbabel [Citation28]. Unavailable structures on the database were prepared with ChemDraw 19.0 and converted to the mol2 format. Gasteiger-type polar hydrogen charges were attributed while the nonpolar hydrogen atoms merged with the carbons. The Universal Force Field (UFF) was employed as the energy minimization parameter. The ligand and protein molecules were further converted to dockable pdbqt format using Autodock tools.
Molecular docking Study
Active site targeted molecular docking of phytochemicals
After a blind docking of the OGDPs and reference inhibitors against the APB2P targets, an active site directed docking of the hit OGDPs from the blind docking analysis and reference compounds to the APB2Ps was executed with AutoDock Vina incorporated in PyRx 0.8 [Citation29]. The OGDPs were imported and minimized with OpenBabel in PyRx 0.8 [Citation28]. The conjugate gradient descent was applied as the optimization algorithms, while the Universal Force Field (UFF) was employed as the energy minimization parameter. For the docking studies, the catalytic pocket of the APB2Ps defined by a grid box size and center (Table S1) were determined by drawing the box around the catalytic residues that are involved in the interaction with the native ligands was employed. Default mode was used for other parameters.
Molecular dynamics
Molecular Dynamic Simulation utilizing Nanoscale Molecular Dynamics (NAMD) V2.13 [Citation30] was performed on 3ZLR (unbound), 3ZLR-luteolin complex, 4LVT (unbound) and 4LVT-ursolic acid complex. Prior to the MDS analysis, necessary files were prepared using CHARMM-GUI webserver [Citation31,Citation32]. The water model TIP3P was used in the solvation while the physiological concentration of 0.154 M was set by neutralized NaCl ions. The systems were minimized for 10,000 steps using CHARMM 36 force field as conjugate gradient algorithm in constant number of atoms, volume, and temperature ensemble (NVT). System equilibration was started afterward for 1 ns in NPT (constant number of atoms, constant pressure, and constant temperature ensemble). A 310 K temperature was set and maintained by langevin dynamics, while A 1 atm pressure was set and maintained by Nose-Hoover langevin piston. Eventually, All the systems were run for 100 ns in NVT ensemble. Periodic Boundary Conditions (PBC) were applied throughout the simulation. The trajectories were saved for each 0.1 ns with a 2 femto second time step. VMD scripts were used to analyze the trajectories by calculating the various thermodynamic parameters [Citation33].
Binding Free energy calculations
The free binding energy was computed employing the Molecular Mechanics Generalized Born Surface Area (MMGBSA) applied in MMPBSA.py in Ambertools 17 package. The method of generalized born (igb) and saltcon variable was set to 5 and 0154 M respectively. The contribution of amino acid residues to the computed binding free energy was decomposed [Citation34]. The clustering of the MDS trajectories was done using the elbow method with TTClust V 4.9.0 [Citation35].
ADMET study
The lead phytochemicals to the APB2P were evaluated for drug-likeness using Lipinski filtering tool analysis [Citation36]. The ADMET study was analyzed on a SuperPred webserver (http://lmmd.ecust.edu.cn/admetsar1/predict/) [Citation37] (https://admet.scbdd.com/calcpre/index_sys_result/) [Citation38]. The tool utilizes important molecular descriptors for the ADMET filtering, and important physiochemical properties of the ligands for the drug likeness prediction. This tool was employed for predicting crucial descriptors of drug likeness.
Results
Molecular docking
The docking scores of the 103 OGDPs and the reference compounds (navitoclax, WEHI-539 and gambogic acid) against the target proteins are represented in Table S2. A hit-list comprising the top twenty ranked phytochemicals from OGDPs was defined based on the docking scores and interactions with important catalytic residues of the APB2Ps (Table S3). From the interaction of the hit phytocompounds, the two lead OGDPs with lowest binding energies for each of the targets were compiled, affording a combined list of five lead phytochemicals (Ursolic acid, β-sitosterol, luteolin, apigenin 7,4’,dimethyl ether, basilimoside). Among these five phytochemicals, high binding tendencies to multiple APB2Ps were observed. Ursolic acid, with the top ranked binding energy (−8.3 Kcal/mol) for BCL-2, exhibited highest binding affinities and interactions with multiple APB2P viz: Mcl-1 (−9.8 Kcal/mol), Bfl-1 (−8.1 Kcal/mol) and Bcl A1 (−7.8 Kcal/mol. Luteolin interacted with the topmost binding affinity with BCL-XL (−9.8 Kcal/mol) and the second top ranked phytochemical to the Mcl-1 (−9.4 Kcal/mol). Ursolic acid had a close binding energy to Bcl-2 as gambogic acid and WEHI_539. In the case of BCL-XL protein, luteolin, and apigenin 7,4’-dimethyl ether had the highest binding energies. Similarly, Ursolic acid, β-sitosterol, calamenene, luteolin, γ-cadinene, δ-cadinene, apigenin with −9.8, −9.4, −9.1, −9, −8.9, −8.9, −8.7 Kcal/mol respectively exhibit greater tendency of high affinity binding to MCL-1 than that of WEHI-539 (with −8.6 Kcal/mol) (Table S2). Basilimoside with binding energy of −8.8 Kcal/mol exhibit higher binding affinity with BFL-1 than WEHI-539 (−8.4 kcal.mol1) and Gambogic acid (−7.6 Kcal/mol), Ursolic acid, apigenin 7,4’-dimethyl ether, apigenin have higher binding tendency than Gambogic acid. Overall, Ursolic acid, β-sitosterol, luteolin, exhibit remarkable high affinity binding with multiple anti-apoptotic proteins ().
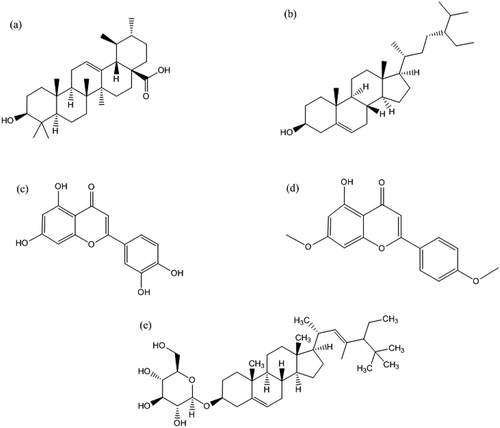
Figure 1. Chemical structure of potential inhibitors of anti-apoptotic BCL-2 proteins (a) ursolic acid, (b) β -Sitosterol, (c) luteolin, (d) apigenin 7,4’-dimethyl ether, and (e) basilimoside.
Amino acid interaction of selected bioactive compounds with anti-apoptotic proteins
The amino acid interaction of the lead phytochemicals (Ursolic acid, β-sitosterol, luteolin, apigenin 7,4’,dimethyl ether, basilimoside) and reference inhibitors to the amino acid residues of five APB2Ps are reported in .
Table 1. Interacting amino acid residue of anti-apoptotic BCL-2 proteins with top docked compounds.
The docking revalidation of the co-crystallized inhibitor (navitoclax) with Bcl-2 presented navitoclax to be well docked into to BH3 binding groove of Bcl-2. Navitoclax stretched into the P2 and P4 binding pocket of the groove. In a similar binding mode as navitoclax the best docked OGDPs to Bcl-2 (Ursolic acid) were oriented in the BH3 binding pocket. The 1, 6a, 9 and 12-methyl groups attached to the pentacyclic rings of ursolic acid formed alkyl contact with Tyr105, Tyr105, Arg143 and Arg104 respectively, the B and C rings formed Pi-Alkyl contact with Tyr105. Residue Gly142 of the Bcl-2 formed H-bond with the carbonyl oxygen of ursolic acid ().
Figure 2. Details of binding mode (a) solvent-accessible surface view (b) interaction view of ligands in BH3 binding pocket of Bcl-2. Stick representations of the Ligands are shown by colors (bi) blue: β-Sistosterol (bii) red: Ursolic acid (biii) green: navitoclax (referencer inhibitors). : Types of interactions are represented by Green-dotted lines: H-bonds; light purple-dotted line: hydrophobic interactions (Pi-Alkyl, Alkyl and pi-stacking); purple-dotted line: Pi-Pi T Shaped; yellow-dotted lines: Pi-sulfur interactions, pi-stacking interactions, with three-letter abbreviations of amino acids.
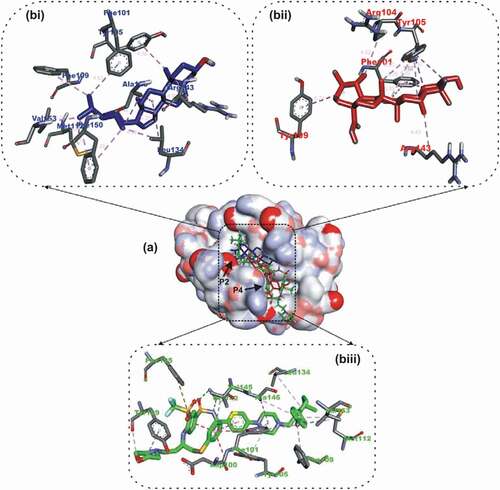
The B ring of the beta-sitosterol formed Pi-Alkyl interaction with Arg143 while the C ring interacted via Pi-alkyl contact to Ala146 and Leu134. Three alkyl contacts were observed between the C24’ of ursolic acid and residues: Phe150, Leu134 and Ala146. The methyl end of C26 formed alkyl contacts with Met112, PHE150 and Val153. C27 formed alkyl contacts with phenyl alanine residues (Phe109 and Phe101) of BCL-2. Luteolin, the top docking compound with the BCL-XL APB2Ps was docked into the BH3 binding groove of BCL-XL in the same orientation as the native ligand WEHI_539 ().
Figure 3. Details of binding mode (a) solvent-accessible surface view (b) interaction view of ligands in BH3 binding pocket of Bcl- XL. Stick representations of the Ligands are shown by colors (bi) blue: Luteolin (bii) red: Apigenin 7,4’,dimethyl ether (biii) green: WEHI_539 (referencer inhibitors).
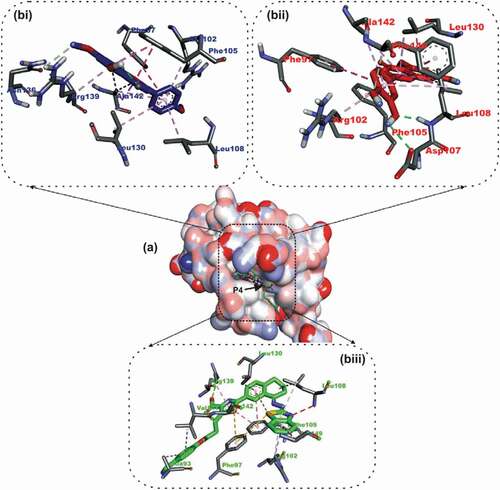
Both compounds were oriented in the BCL-XL hydrophobic groove, showing remarkable interaction with Phe105 in the P2 binding pocket. The A ring of luteolin formed 2 Pi-Alkyl interactions and Pi-Pi Stacking with Leu130, Phe105 and Phe105 of BCL-XL respectively (). The C ring also formed 2 Pi-Alkyl interactions, a Pi-Pi T-shaped and Pi-Sigma contact with Ala142, Leu108, Phe105 and Leu130 respectively. Three Pi-Alkyl interactions, and 2 Pi-Pi T shaped contacts were made by the B ring of luteolin to Arg102, Ala142, Leu108, Phe97 and Phe146. The 4’-hydroxyl and 5’-hydroxyl groups linked to the B ring formed 2 and a hydrogen bond With LEU108, ASP107 and Phe146 respectively ().
In a similar binding as the co-crystallized native ligand (2-Indole-acylsulfonamide) with Mcl-1, gambogic acid, a known inhibitor of Mcl-2, ursolic acid the top docked OGDPs to Mcl-2 were oriented into the hydrophobic P4 pocket. The 1,2, 6a and 9- methyl groups of ursolic acid interacted with alkyl contacts to Met250, Val253, Ala227 And Met231 ().
Figure 4. Details of binding mode (a) solvent-accessible surface view (b) interaction view of ligands in BH3 binding pocket of Mcl-1. Stick representations of the Ligands are shown by colors (bi) blue: Ursolic acid (bii) red: luteolin (biii) green: Gambogic Acid (referencer inhibitors).
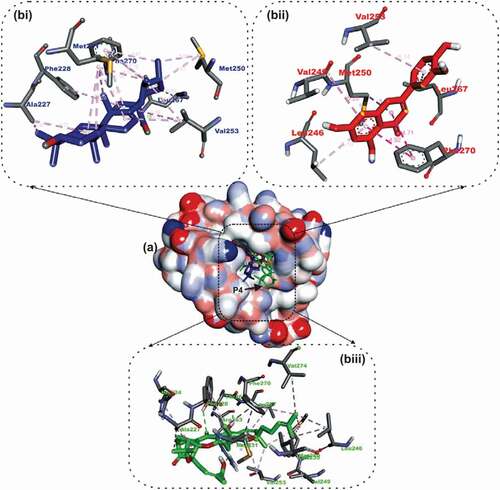
The B Ring formed Pi-Alkyl contact with Ala227 and Met231, while the carbonyl oxygen formed hydrogen bond with Leu267. Ursolic acid also double as the lead OGDPS to the Bfl-1 and Bcl A1 APB2Ps. Ursolic acid was docked into the BH3 hydrophobic pocket of the two APB2Ps ().
Figure 5. Details of binding mode (a) solvent-accessible surface view (b) interaction view of ligands in BH3 binding pocket of Bfl-1. Stick representations of the Ligands are shown by colors (bi) blue: ursolic acid (bii) red: basilimoside.
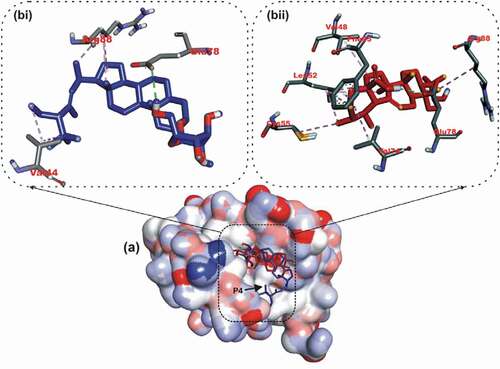
Figure 6. Details of binding mode (a) solvent-accessible surface view (b) interaction view of ligands in BH3 binding pocket of Bcl-A1. Stick representations of the Ligands are shown by colors (bi) blue: ursolic acid (bii) red: β – Sitosterol.
The energy profile of lead phytochemicals to respective proteins is represented in the (Figures S6- 115 Supplementary Data)
Molecular dynamic simulations of top complexes
The conformational stability of the unbound Bcl-2 and BCL-XL compared with the complexes formed with the lead OGDPs was monitored through MD simulation. The VMD Tk console scripts were used for the analysis of the MDS trajectories for the calculation of the thermodynamic parameters viz: SASA, RoG, RMSD, RMSF and number of H-bonds. SASA and RoG values estimate the changes in the protein folding. Increase in both parameters is an indication that the protein is unfolded during the simulation and vice versa. The Apo protein (unbound) BCL-XL shows an initial overall decrease in SASA () and RoG ().
Figure 7. The Surface Accessible Surface Area (SASA) plots of molecular dynamics (MD) simulation of anti-apoptotic BCL-2 proteins (APB2P) (A) BCL-XL (B) BCL-2. Blue line: Apo protein, Pale Orange line: Complexes of APB2P with top dock phytochemicals (A: Luteolin and B: Ursolic acid).
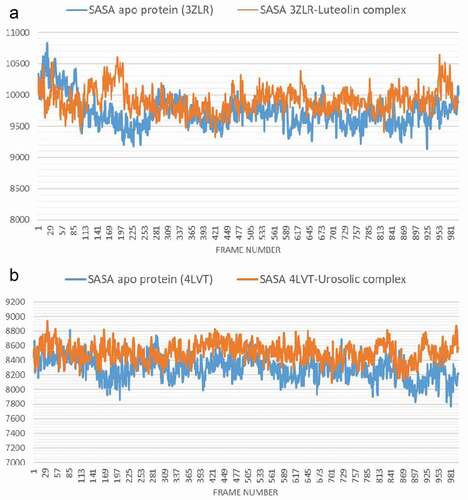
Figure 8. The Radius of gyration (RoG) plots of molecular dynamics (MD) simulation of anti-apoptotic BCL-2 proteins (APB2P) (A) BCL-XL (B) BCL-2. Blue line: Apo protein, Pale Orange line: Complexes of APB2P with top dock phytochemicals (A: Luteolin and B: Ursolic acid).
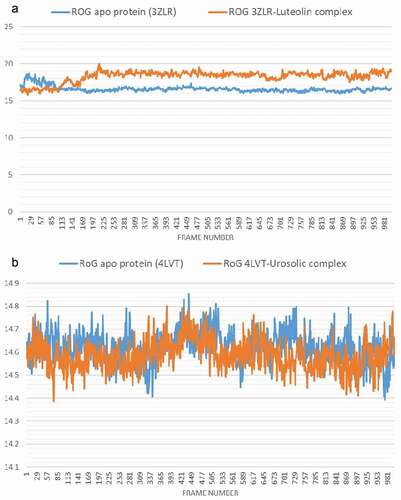
Which corresponds to the rotation of amino acids (S1:G18) about 180° and their motion toward the rest of the protein (Figure S16: supplementary data) resulting in a more compact form, however, in the case of BCL-XL -luteolin, the Figure shows that the amino acids (S1:G18) have moved away from the protein increasing its surface area, which is confirmed by the RoG values. The SASA plot shows a steady fluctuation around average values of 8292 Å2 and 8508 Å2 for the unbound BCL-2 () and BCL-2 -ursolic acid complex (), indicating no conformational changes along the trajectories. However, the unbound BCL-2 shows a decrease between 14 ns to 24 ns, corresponding to the motion of the amino acids (A41:Q59) (Figure S11: supplementary data) close to the rest of the protein closing the pocket. This can be seen from the RMSD plots () which shows a deviation during the same time period which is not seen in the BCL-2-ursolic acid complex due to the presence of the ursolic acid in the pocket. The RoG plots for the two systems fluctuated around 14.62 Å and 14.58 Å for the unbound BCL-2 () and BCL-2 -Ursolic acid complex ().
Figure 9. The Backbone-Root Mean Square Deviation (RMSD) plots of molecular dynamics (MD) simulation of anti-apoptotic BCL-2 proteins (APB2P) (A) BCL-XL (B) BCL-2. Blue line: Apo protein, Pale Orange line: Complexes of APB2P with top dock phytochemicals (A: Luteolin and B: Ursolic acid).
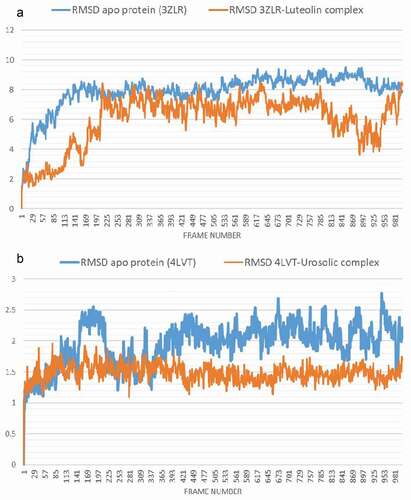
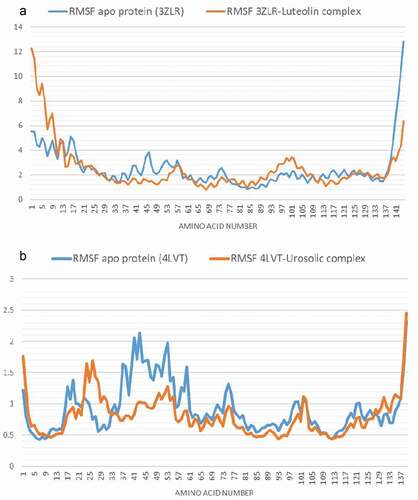
The RMSD plot depicts how the frames have deviated from the initial structure. Both the unbound BCL-XL and the BCL-XL -luteolin complex () equilibrated after nearly 15 and 22 ns, respectively with mean values of 7.8 Å and 5.9 Å, respectively. The RMSF plot shows on average, how much each amino acid has deviated from the initial pose. The large values at both the beginning and the end of the simulation correspond to the fluctuation at the N- and C-terminals ( and )
Figure 10. Per residue Root Mean Square Fluctuations (RMSF) plots of molecular dynamics (MD) simulation of anti-apoptotic BCL-2 proteins (APB2P) (A) BCL-XL (B) BCL-2. Blue line: Apo protein, Pale Orange line: Complexes of APB2P with top dock phytochemicals (A: Luteolin and B: Ursolic acid).
Both the unbound BCL-XL and BCL-XL -luteolin complex have an average number of H bonds of 33 ().
Figure 11. The changes in the number of H-bonds during the MDS trajectory of anti-apoptotic BCL-2 proteins (APB2P) (A) BCL-XL (B) BCL-2. Blue line: Apo protein, Pale Orange line: Complexes of APB2P with top dock phytochemicals (A: Luteolin and B: Ursolic acid).
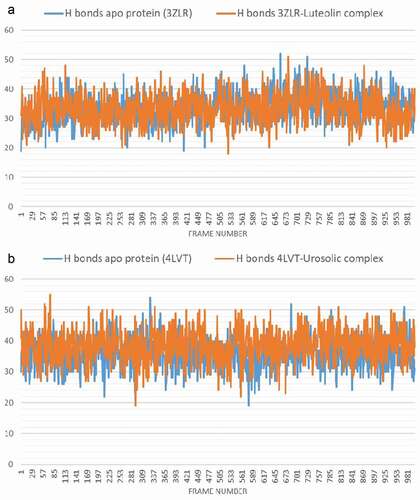
The average H bonds for the unbound BCL-2 and BCL-2 -ursolic acid complex () are 36 and 38 respectively, the binding of ursolic acid may have increased the intramolecular hydrogen bonds leading to a more compact structure as shown by the RMSD () and RMSF () plots.
Binding Free Energy Calculation
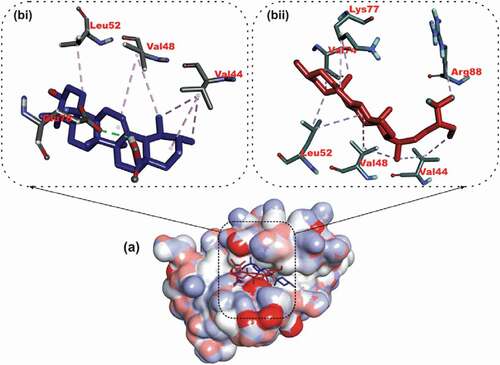
The binding free energy computed using MMGBSA for BCL-XL-luteolin and BCL-2- Ursolic acid is−25.1064 and −21.19 Kcal/mol. The decompositional analysis of the binding free energy shows the key binding amino acids contributing to the binding of luteolin. Luteolin interacted strongly with Phe38, Tyr42, Arg43, Phe46, Leu49, Leu71 and Arg80 which correspond to the amino acid residue number (Phe97, Tyr102, Phe105, Leu108, Leu130 and Arg139) as numbered in the experimental 3-dimensional structure ().
Figure 12. Molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) plot of binding free energy contribution per residue of (A) BCL-XL – Luteolin complex and (B) BCL-2- Ursolic acid complex.
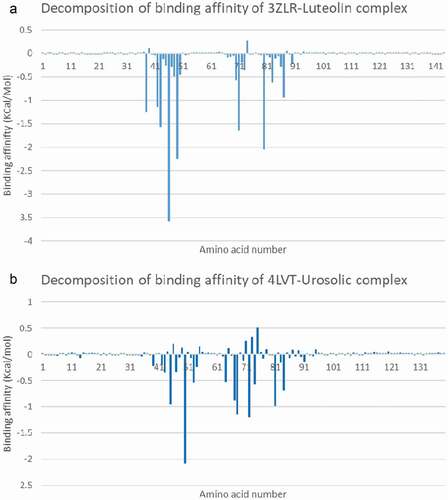
Ursolic acid interacted with Arg45, Phe47, Met50, Gln53, Leu54, Arg64, Thr67, Val68, Leu72, Arg81, and Alal84, which also correspond to (Arg107, Phe110 Met112, Gln115, Leu116, Arg126, Thr129, Val130, Leu134, Arg143 and Ala146) () as numbered in the deposited crystal structures.
Cluster Analysis
Analysis of the clusters shows that interactions with Phe46 and Arg43 residues were preserved in the BCL-XL-luteolin complex. While Asp46 in BCL-2-Ursolic acid complex was preserved in the clusters. show interactive view of the first and last cluster from both systems while the full amino acid interaction in all clusters are presented in Table S11: supplementary data.
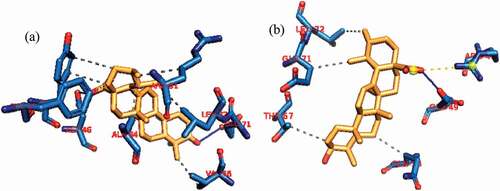
Figure 13. Interactions in human BCL-XL -Luteolin complex (a) cluster 1 and (b) cluster 4. Grey dotted dashed lines: hydrophobic interactions; Blue solid line: Hydrogen bond. Amino acids are in one-letter format and blue color. Luteolin is in Orange stick representation.
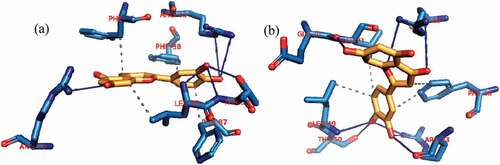
Figure 14. Interactions in human BCL-2-ursolic acid complex (a) cluster 1 and (b) cluster 4. Grey dotted dashed lines: hydrophobic interactions; Blue solid line: Hydrogen bond. Amino acids are in one-letter format and blue color. Ursolic acid is in Orange stick representation.
Physicochemical properties of high affinity compounds
The five lead phytochemicals (ursolic acid, β -Sitosterol, Luteolin, Apigenin 7,4’-dimethyl ether, and Basilimoside) with high binding energy to the APB2Ps were examined for drug-likeness and ADMET properties.
The results for the ADMET analyses of lead phytochemicals are presented in Table S5: supplementary data. The five-lead presented favorable drugable and ADMET properties over a wide range of physiochemical and molecular descriptors. They were also predicted to be non-AMES toxic and non-carcinogenic.
Discussion
The key regulators of apoptosis in the cell such as the BCL- 2 proteins have been explored as drug targets towards developing anticancer drugs [Citation39]. Presently, more than 20 members of the family have been discovered [Citation40]. Among them, the anti-apoptotic proteins comprising all four BH domains viz BCL-2, BCL-W, BCL-XL, MCL-1, BCL-B BFL-1, etc. inhibit apoptosis by sequestering multidomain pro-apoptotic members such as BAX and BAK or by suppressing activator BH3-only proteins such as BIM, PUMA, BID, and NOXA. A delicate equilibrium is sustained between the pro-and anti-apoptotic proteins, while increase of pro-apoptotic proteins results in continual apoptosis, repletion of the anti-apoptotic proteins circumvents the apoptotic pathway, by inhibiting other pro-apoptotic proteins. Any interruption of this equilibrium is implicated in cancer and other affiliated diseases [Citation41]. In the present study, we explored the interactions of 103 O. gratissimum-derived phytochemicals (OGDPs) against five APB2Ps employing computational and biophysical methods. From the result of the static docking calculations five OGDPs (ursolic acid (UA), β -bitosterol, luteolin, apigenin 7,4’-dimethyl ether, and basilimoside) exhibit high affinity binding to the targets proteins. These phytochemicals were docked into the BH3 binding grooves of the APB2Ps. The binding mode analysis showed that five lead OGDPs were oriented in the active sites, interacting with catalytic residues of the APB2Ps in a binding mode similar to that of the reference inhibitors [Citation42]. This is in tandem with a similar study by Adewole and Kayode who reported that, Morinda lucida-derived ursolic acid and oleanolic acid and other phytosterols had high binding energies for APB2Ps [Citation4]. The observed minimal binding energy with the catalytic residues of the APB2Ps may have resulted from the hydrophobic pockets nature of the pockets, since the phytochemicals had groups that can interactive with hydrophobic residues [Citation43].
Among the 5 compounds with lowest binding energy, UA, β–Sitosterol and luteolin exhibited high affinity binding to multiple anti-apoptotic proteins. The beneficial effects of UA, which include anti-oxidant, anti-apoptotic, anti-inflammatory, and anti-carcinogenic have attracted special interest [Citation44]. The higher binding tendencies for BCL-2 and BFL-1 by ursolic acid than Gambogic acid is in tandem with several in vitro studies that reports their modulatory effects on this protein. For instance, early report shows that, treatment with ursolic acid led to the down regulation of BCL-2 protein in MCF-7 breast cancer cells [Citation45]. The modulatory effects of β-sitosterol and other phytosterols on the activity of these proteins have been studied in vitro. For instance, phytosterols isolated from the root extracts of Clinacanthus nutans inhibited BCL-2 in MCF-7 cells [Citation46]. Specifically β-sitosterol induced significant reduction of Bcl-2 proteins in MCA-102 fibrosarcoma cells [Citation47]. β-sitosterol elevated the ratio of Bax/Bcl-2 in human U937 cells [Citation48]. A recent studies showed that, other phytosterols such as cycloartenol modified the ratio of Bax/Bcl-2 in glioma U87 cells [Citation49]. Another study demonstrated the Bcl-2 proteins expression reducing effect of stigmasterol in human SNU-1 gastric cancer cell line [Citation50]. Luteolin (3,4,5,7-tetrahydroxy flavone) is a dietary flavonoid found in different vegetables, fruits and medicinal plants. The cancer cells inhibitory activity of luteolin against various preclinical models such as human breast, lung, glioblastoma, prostate, pancreatic, and colon cancers has been widely reported [Citation51]. Luteolin has been reported to inhibit carcinogenesis in vitro and in vivo through various mechanisms which include: protection from carcinogenic stimuli, inhibition of proliferative potential of tumor cells, and activation and promotion of cell cycle arrest, and induction of apoptotic process through different signaling pathways [Citation52]. A recent study showed that the proliferation of the breast cancer cell line (MDA-MB-231) was efficiently suppressed by luteolin in a concentration dependent manner [Citation53]. Luteolin has also been reported to enhance mitochondrial membrane potential collapse and opening of the permeability transition pore with the release of cytochrome c through inhibition of Bcl-2 expression [Citation54].
The MD simulation revealed desirable thermodynamics parameters (RMSD, SASA, RMSF, RoG and number of H-bonds) for the top ranked phytochemicals in complexes with the Bcl-2 and BCL-XL APB2Ps. The full 100 ns atomistic MD simulation trajectory files revealed a high stable conformation throughout the simulation when compared to the unbound proteins. The SASA and RoG were evaluated to access the compactness as well as solvent accessibility to the Bcl-2 and BCL-XL APB2Ps complexes with ursolic acid and luteolin respectively. A reasonably steady SASA and RoG were maintained throughout the simulation time, indicating compacted a well folded structure with intact intermolecular [Citation55]. Fewer fluctuations were observed around the interacting residues in the complexes as noticed from the RMSF plots. Previous report has established that increased structural fluctuations usually occur in ligands groups participating in catalysis, notably the substrate binding sites [Citation56], hence the binding of the phytochemicals increased the overall stability of the APB2Ps. The RMSD plots and computed averages proved that the binding of UA and luteolin to the active regions of Bcl-2 and BCL-XL did not alter the structural conformation of the protein, the lower RMSD values of the complex systems indicates a more compact structure upon the binding of the phytochemicals [Citation57]. The close number of H-bonds between the unbound APB2Ps protein and complex systems, this further shows that the structural conformation of the protein was not altered in the simulated systems [Citation57]. Binding free energies computed from MD simulation trajectories is known to provide more precise estimate of free energies, than those gotten from static molecular docking calculations [Citation58]. Thus, the binding free energies are essential to gain in-depth insight into the binding interactions of the lead compounds in drug design [Citation59]. The results from the computation of free energy of binding decomposition by interacting residues of BCL-XL and BCL-2 with luteolin and ursolic acid respectively shows that the phytochemicals maintained the interactions with active site residues in the dynamic simulation environment further indicating the strong binding potential of the luteolin and ursolic acid to the respectively proteins. Clustering of the MD simulation trajectories of the complex systems established that the interactions (H-bonds and hydrophobic interaction) in the representative structures of the clusters were preserved at different time frames, further suggesting that the binding interactions reported in this study can be sustained for experimental procedures.
The findings from drug-likeness and ADMET analysis showed that all the five lead phytochemicals (ursolic acid, β -Sitosterol, Luteolin, Apigenin 7,4’-dimethyl ether, and Basilimoside) to the BCL-2 proteins, satisfied the requisite parameters used by the Lipinski rule of five filtering tool. This suggests favorable physicochemical/druggable properties of the compounds [Citation60]. Most importantly the aqueous solubility values for these compounds were less than zero, indicating that all the five compounds are qualified to be used as drugs. Since the drug likeness of a given compound is correlated to its solubility in aqueous medium this further gave a positive indication to their bioavailability [Citation61]. The 5 lead phytochemicals demonstrated high probability gastrointestinal absorption with high bioavailability [Citation62]. The permeability glycoprotein (P-gp) is expressed on the surfaces of several cells saddled with the role of pumping back compounds or xenobiotics into the intestinal lumen, and other parts of the body such as capillaries and urine-conducting ducts and [Citation63]. All the lead phytochemicals depicted non-substrate probability of the P-gp. These property further enhances the bioavailability and reduced rate of excretion for the phytochemicals, hence the high their half-lives [Citation63]. The function of the hERG channel in cell repolarization and the termination of action potential of cardiac cell is known, compounds that block the channel may result in cardiotoxicity [Citation64]. The five lead phytochemicals presented low probability of blocking the hERG channel, indicating their inability of causing cardiotoxicity that are related to hERG channel [Citation64]. The five lead phytochemicals did not display AMES mutagenicity, human hepatotoxicity, may not induce liver injury for drug toxicity and may not cause skin sensitization All these descriptors shows that the lead phytochemicals may not cause genetic mutations which is associated with the pathophysiology of diseases, like cancer and hepatoxicity [Citation65] Using several cytochrome P450 descriptors, the probability for the lead phytochemicals to affect phase I drug metabolism in the liver was also studied. The various cytochrome P450 were not inhibited by lead phytochemicals, hence they may not contrary affect the phase I drug metabolism in the liver [Citation66] These phytochemicals may be therefore regarded to be non toxic with well-disposed druggable properties. Although the results from the predicted acute toxicity showed that ursolic acid, β -Sitosterol and basilimoside presented moderate acute toxicity, while luteolin and pigenin 7,4’,dimethyl ether had low acute toxicity [Citation67,Citation68], previous experimental studies have shown that β -Sitosterol [Citation48], ursolic acid [Citation45] inhibited this proteins at an appreciable low doses in an in vitro studies, therefore they can be subjected for further in vivo experimental studies.
Conclusion
Over expression of anti-apoptotic Bcl-2 family proteins is associated with different types of cancer. The anti-cancer activities of Ocimum gratissimum have been reported but the anti-cancer constituent(s) have not been identified. Herein, we applied different computation techniques to screen 103 Ocimum gratissimum derived phytochemicals for potential inhibitors against BCL-2, MCL-1, BCL-B BCL-XL and BFL-1. The Results from this study showed that five lead phytochemicals (ursolic acid, β-sitosterol, luteolin, apigenin 7,4’,dimethyl ether, basilimoside) had high binding tendencies to the five APB2Ps. Ursolic acid, β-sitosterol and luteolin exhibited different levels of multiplicity binding to atleast two of the anti-apoptotic Bcl-2 proteins which may in turn exert inhibitory effect to these proteins. The five lead phytochemicals satisfied the requisite properties for the in silico drug likeness analysis with well favorable ADMET properties. Ursolic acid and luteolin, complexed with the Bcl-2 and BCL-XL respectively, demonstrated high flexibility and structural stability in the 100 ns MD simulation. Evidences from previous work has established the inhibitory potential of ursolic acid, β-sitosterol and luteolin against the APB2Ps. The results suggest that the anti-proliferative activity O. gratissimum may be as a result of the synergistic activities of, at least, the lead phytochemicals. There is therefore further need to investigate these phytochemicals as potential drug candidates.
Competing of interest
The authors declare that they have no competing interests.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Anantram A, Kundaikar H, Degani M, et al. Molecular dynamic simulations on an inhibitor of anti-apoptotic Bcl-2 proteins for insights into its interaction mechanism for anti-cancer activity. J Biomol Struct Dynami. 2019;37(12):3109–3121.
- Petros AM, Medek A, Nettesheim DG, et al. Solution structure of the antiapoptotic protein bcl-2. Proc Nat Acad Sci. 2001;98(6):3012–3017.
- Ku B, Liang C, Jung JU, et al. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21(4):627–641.
- Adewole KE, Ishola AA. Phytosterols and triterpenes from Morinda lucida Benth (Rubiaceae) as potential inhibitors of anti-apoptotic BCL-XL, BCL-2, and MCL-1: an in-silico study. J Recept Signal Transduct. 2019;39(1):87–97.
- Wilson WH, O’Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–1159.
- Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208.
- Ashkenazi A, Fairbrother WJ, Leverson JD, et al. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–284.
- Nweze EI, Eze EE. Justification for the use of Ocimum gratissimum L in herbal medicine and its interaction with disc antibiotics. BMC Complement Altern Med. 2009;9(1):1–6.
- Ojo OA, Ojo AB, Oyinloye BE, et al. Ocimum gratissimum Linn. Leaves reduce the key enzymes activities relevant to erectile dysfunction in isolated penile and testicular tissues of rats. BMC Complement Altern Med. 2019;19(1):1–10.
- Clark J, You M. Chemoprevention of lung cancer by tea. Mol Nutr Food Res. 2006;50(2):144–151.
- Priyanka C, Shivika S, Vikas S. Ocimum gratissimum: a review on ethnomedicinal properties, phytochemical constituents, and pharmacological profile. In: Kumar N, editor. Biotechnological Approaches for Medicinal and Aromatic Plants: conservation, Genetic Improvement and Utilization. Springer Singapore: Singapore; 2018. p. 251–270.
- Ekunwe SI, Thomas MS, Luo X, et al. Potential cancer-fighting Ocimum gratissimum (OG) leaf extracts: increased anti-proliferation activity of partially purified fractions and their spectral fingerprints. Ethn Dis. 2010;20(1):12.
- Nangia-Makker P, Raz T, Tait L, et al. Ocimum gratissimum retards breast cancer growth and progression and is a natural inhibitor of matrix metalloproteases. Cancer Biol Ther. 2013;14(5):417–427.
- Huang -C-C, Hwang J-M, Tsai J-H, et al. Aqueous Ocimum gratissimum extract induces cell apoptosis in human hepatocellular carcinoma cells. Int J Med Sci. 2020;17(3):338.
- Gupta S, Prakash J, Srivastava S. Validation of traditional claim of Tulsi, Ocimum sanctum Linn. as a medicinal plant; 2002.
- Do Nascimento Silva MK, Carvalho VRDA, Matias EFF. Chemical profile of essential oil of Ocimum gratissimum L. and evaluation of antibacterial and drug resistance-modifying activity by gaseous contact method. Pharmacogn J. 2016;8(1):19.
- Selvaraju R, Sakuntala P, Jaleeli KA. GC–MS and FTIR Analysis of Chemical Compounds in Ocimum Gratissimum Plant. Biophysics. 2021;66(3):401–408.
- Matasyoh LG, Matasyoh JC, Wachira FN, et al. Chemical composition and antimicrobial activity of the essential oil of Ocimum gratissimum L. growing in Eastern Kenya. Afr J Biotechnol. 2007;6(6).5.
- Kpadonou Kpoviessi BG, Ladekan EY, Kpoviessi DSS, et al. Chemical variation of essential oil constituents of Ocimum gratissimum L. from Benin, and impact on antimicrobial properties and toxicity against Artemia salina Leach. Chem Biodivers. 2012;9(1):139–150.
- Fokou JBH, Dongmo PM, Boyom FF, et al. Antioxidant and antifungal activities of the essential oils of Ocimum gratissimum from Yaoundé and Dschang (Cameroon). J Pharm Pharmacol. 2014;2:257–268.
- Melo RS, Albuquerque Azevedo ÁM, Gomes Pereira AM, et al. Chemical composition and antimicrobial effectiveness of Ocimum gratissimum L. essential oil against multidrug-resistant isolates of Staphylococcus aureus and Escherichia coli. Molecules. 2019;24(21):3864.
- Runyoro D, Ngassapa O, Vagionas K, et al. Chemical composition and antimicrobial activity of the essential oils of four Ocimum species growing in Tanzania. Food Chem. 2010;119(1):311–316.
- Joshi R. Chemical composition, in vitro antimicrobial and antioxidant activities of the essential oils of Ocimum gratissimum, O. sanctum and their major constituents. Indian J Pharm Sci. 2013;75(4):457.
- Katara A, Pradhan CK, Singh P, et al. Volatile Constituents and Antimicrobial Activity of Aerial parts of Ocimum gratissimum Linn. J Essential Oil Bearing Plant. 2013;16(2):283–288.
- Prabhu KS, Lobo R, Shirwaikar AA, et al. Ocimum gratissimum: a review of its chemical, pharmacological and ethnomedicinal properties. Open Complement Med J. 2009;1(1):1–15.
- Monga S, Dhanwal P, Kumar R, et al. Pharmacological and physico-chemical properties of Tulsi (Ocimum gratissimum L.): an updated review. Pharma Innovation. 2017;6(4, Part C):181.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- O’Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3(1):33.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802.
- Lee J, Cheng X, Swails JM, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput. 2016;12(1):405–413.
- Brooks BR, Brooks CL, Mackerell AD, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38.
- Suomivuori C-M, Latorraca NR, Wingler LM, et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science. 2020;367(6480):881–887.
- Salentin S, Schreiber S, Haupt VJ, et al. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447.
- Nickel J, Gohlke B-O, Erehman J, et al. SuperPred: update on drug classification and target prediction. Nucleic Acids Res. 2014;42(W1):W26–W31.
- Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105.
- Dong J, Wang -N-N, Yao Z-J, et al. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. 2018;10(1):1–11.
- Shamas-Din A, Kale J, Leber B, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714.
- Timucin AC, Basaga H, Kutuk O. Selective targeting of antiapoptotic BCL‐2 proteins in cancer. Med Res Rev. 2019;39(1):146–175.
- Ramos J, Muthukumaran J, Freire F, et al. Shedding light on the interaction of human anti-apoptotic Bcl-2 protein with ligands through biophysical and in silico studies. Int J Mol Sci. 2019;20(4):860.
- Sleebs BE, Kersten WJA, Kulasegaram S, et al. Discovery of Potent and Selective Benzothiazole Hydrazone Inhibitors of Bcl-X L. J Med Chem. 2013;56(13):5514–5540.
- Priya P, Maity A, Majumdar S, et al. Interactions between Bcl-xl and its inhibitors: insights into ligand design from molecular dynamics simulation. J Mol Graphics Modell. 2015;59:1–13.
- Seo DY, Lee SR, Heo J-W, et al. Ursolic acid in health and disease. Korean J Physiol Pharmacol. 2018;22(3):235.
- Kassi E, Sourlingas TG, Spiliotaki M, et al. Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Invest. 2009;27(7):723–733.
- Teoh PL, Cheng AYF, Liau M, et al. Chemical composition and cytotoxic properties of Clinacanthus nutans root extracts. Pharm Biol. 2017;55(1):394–401.
- Moon D-O, Lee K-J, Choi YH, et al. β-Sitosterol-induced-apoptosis is mediated by the activation of ERK and the downregulation of Akt in MCA-102 murine fibrosarcoma cells. Int Immunopharmacol. 2007;7(8):1044–1053.
- Park C, Moon D-O, Rhu C-H, et al. β-Sitosterol induces anti-proliferation and apoptosis in human leukemic U937 cells through activation of caspase-3 and induction of Bax/Bcl-2 ratio. Biol Pharm Bull. 2007;30(7):1317–1323.
- Niu H, Li X, Yang A, et al. Cycloartenol exerts anti-proliferative effects on Glioma U87 cells via induction of cell cycle arrest and p38 MAPK-mediated apoptosis. J BUON. 2018;23. 1840–1845.
- Li K, Yuan D, Yan R, et al. Stigmasterol exhibits potent antitumor effects in human gastric cancer cells mediated via inhibition of cell migration, cell cycle arrest, mitochondrial mediated apoptosis and inhibition of JAK/STAT signalling pathway. J BUON. 2018;23(5): 1420–1425.
- Ganai SA, Sheikh FA, Baba ZA, et al. Anticancer activity of the plant flavonoid luteolin against preclinical models of various cancers and insights on different signalling mechanisms modulated. Phytother Res. 2021;35(7):3509–3532.
- Imran M, Rauf A, Abu-Izneid T, et al. Luteolin, a flavonoid, as an anticancer agent: a review. Biomed Pharmacother. 2019;112:108612.
- Huang L, Jin K, Lan H. Luteolin inhibits cell cycle progression and induces apoptosis of breast cancer cells through downregulation of human telomerase reverse transcriptase. Oncol Lett. 2019;17(4):3842–3850.
- Cook MT, Mafuvadze B, BESCH-WILLIFORD C, et al. Luteolin suppresses development of medroxyprogesterone acetate-accelerated 7, 12-dimethylbenz (a) anthracene-induced mammary tumors in Sprague-Dawley rats. Oncol Rep. 2016;35(2):825–832.
- Sinha S, Wang SM. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput Struct Biotechnol J. 2020;18:723–736.
- Dong Y-W, Liao M-L, Meng X-L, et al. Structural flexibility and protein adaptation to temperature: molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc Nat Acad Sci. 2018;115(6):1274–1279.
- Cheng X, Ivanov I. Molecular dynamics. Comput Toxicol. 2012;2012:243–285.
- Perez A, Morrone JA, Simmerling C, et al. Advances in free-energy-based simulations of protein folding and ligand binding. Curr Opin Struct Biol. 2016;36:25–31.
- Kollman PA, Massova I, Reyes C, et al. Calculating Structures and Free Energies of Complex Molecules: combining Molecular Mechanics and Continuum Models. Acc Chem Res. 2000;33(12):889–897.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25.
- Elipilla P. Designing, molecular docking and toxicity studies of novel plasmepsin II inhibitors. Eur J Biotechnol Biosci. 2015;3:27–30.
- Wang -N-N, Huang C, Dong J, et al. Predicting human intestinal absorption with modified random forest approach: a comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues. RSC Adv. 2017;7(31):19007–19018.
- Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics. Clin Pharmacokinet. 2003;42(1):59–98.
- Kratz JM, Grienke U, Scheel O, et al. Natural products modulating the hERG channel: heartaches and hope. Nat Prod Rep. 2017;34(8):957–980.
- Mulliner D, Schmidt F, Stolte M, et al. Computational models for human and animal hepatotoxicity with a global application scope. Chem Res Toxicol. 2016;29(5):757–767.
- Rostkowski M, Spjuth O, Rydberg P. WhichCyp: prediction of cytochromes P450 inhibition. Bioinformatics. 2013;29(16):2051–2052.
- Zhu H, Martin TM, Ye L, et al. Quantitative structure− activity relationship modeling of rat acute toxicity by oral exposure. Chem Res Toxicol. 2009;22(12):1913–1921.
- O’Boyle NM, Sayle RA. Comparing structural fingerprints using a literature-based similarity benchmark. J Cheminform. 2016;8(1):1–14.