
Abstract
A series of rhodium(III) half sandwich complexes of the type [Cp*Rh(PMe3)(S–R–S)]; S–R–S = naphthalene-1,8-dithiolate, acenaphthene-5,6-dithiolate, [1,1’-biphenyl]-2,2’-dithiolate and [2,2’-binaphthalene]-1,1’-dithiolate are reported. In the case of [2,2’-binaphthalene]-1,1’-dithiolate, this represents an infrequent example of a metal complex containing this ligand. All the complexes have been fully characterised using multinuclear NMR spectroscopy and single-crystal X-ray diffraction. The single-crystal X-ray structure of the starting material [Cp*Rh(PMe3)Br2] (1) is also reported for the first time.
Public Interest Statement
S,S′-bidentate ligands remain an important area of chemistry. Complexes bearing this type of ligand have a number of industrial applications including vulcanisation, lubricant additives and catalysis. In addition, S,S′-donors can support unusual magnetic properties and are important in biological systems This work describes the coordination of some simple bidentate S,S ligands to Iridium
1. Introduction
The coordination of S,S′-bidentate ligands remains an important area of chemistry. Complexes bearing this type of ligand have a number of industrial applications including vulcanisation (Bond & Martin, Citation1984; Burns & McAuliffe, Citation1979; Burns, McCullough, & McAuliffe, Citation1980; Eisenberg, Citation2007), lubricant additives (Phillips et al., Citation1995) and catalysis (Bond & Martin, Citation1984; Burns & McAuliffe, Citation1979; Burns et al., Citation1980; Eisenberg, Citation2007). In addition, S,S′-donors can support unusual magnetic properties (Tuna et al., Citation2012; Zhou, Wang, Wang, & Gao, Citation2011) and are important in biological systems (Woollins, Citation1996). As part of our interest in the properties of sulfur donor systems, we have investigated a series of dithiolate ligands bound to aromatic backbones of varying flexibility (Figure ).
Figure 1. Dithiolate ligands studied in this work (charges omitted for clarity).
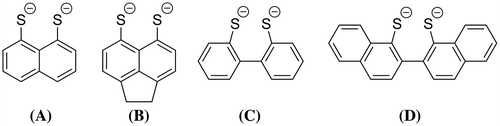
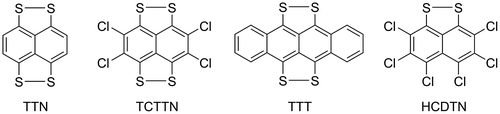
There has been little study on the coordination chemistry of these types of ligands compared to dithiolates such as benzene-1,2-dithiolate or ethane-1,2-dithiolate. One of the most notable exceptions to this was a series of publications by Teo and co-workers in the late 1970s and early 1980s (Teo, Bakirtzis, & Snyder-Robinson, Citation1983; Teo & Snyder-Robinson, Citation1978, Citation1979a, Citation1979b, Citation1981, Citation1984; Teo, Wudl, Hauser, & Kruger, Citation1977; Teo, Wudl, Marshall, & Kruger, Citation1977). They investigated the oxidative addition of the structurally related compounds tetrathionaphthalene (TTN), tetrachlorotetrathionaphthalene (TCTTN) and tetrathiotetracene (TTT) (Figure ) to a variety of low-valent metal centres. With the work focusing on the extensive redox chemistry associated with these “non-innocent” ligands and their potential use as organic solid-state conductors (Teo & Snyder-Robinson, Citation1978, Citation1979a, Citation1979b, Citation1981, Citation1984; Teo, Wudl, Hauser, et al., Citation1977; Teo, Wudl, Marshall, Citation1977, Teo et al., Citation1983). Another interesting system bearing the related hexachlorodithionaphthalene (HCDTN) (Figure ) resulted in an unusual trinuclear nickel complex [Ni3(PPh3)3(S2C10Cl6)3] with the HCDNT acting as a bridging ligand (Bosman & van der Linden, Citation1977).
Figure 2. Structurally related aromatic sulfur donating ligands.
Some of the most recent work involving derivatives of naphthalene-1,8-dithiolate (A) has been in designing [FeFe]-hydrogenase mimics for the production of hydrogen, by both electrochemical and photochemical processes (Figure ) (Figliola, Male, Horswell, & Grainger, Citation2015; Figliola et al., Citation2014; Samuel, Co, Stern, & Wasielewski, Citation2010; Wright, Lim, & Tilley, Citation2009). Complexes involving [1,1’-biphenyl]-2,2’-dithiolate (C) bound to iron have also been investigated as electron transfer catalysts designed to mimic iron hydrogenases (Figure ) (Albers et al., Citation2014; Ballmann, Dechert, Demeshko, & Meyer, Citation2009; Charreteur et al., Citation2010). The coordination chemistry of the structurally related ligand acenaphthene-5,6-dithiolate (B) has received very little investigation out with our own research. (Topf, Monkowius, and Knör (Citation2012) used the acenaphthene backbone as a linker between a 1,2-diimine unit and a dithiolate binding site. The iron carbonyl complex formed using this ligand showed potential as a multielectron transfer photosensitiser for artificial photosynthesis and as a bio-inspired photoredox catalyst (Figure ).
Figure 3. Iron-based catalysts based on A (left), B (centre) and C (right).
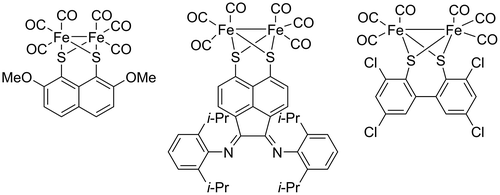
Figure 4. The section of the 1H NMR spectrum of 2c showing the four pseudo triplet of doublets with a splitting diagram showing how these signals are formed.
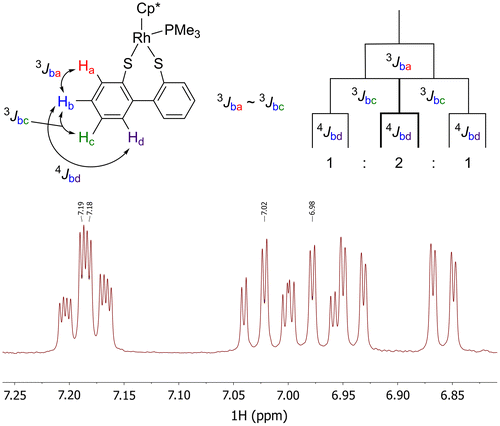
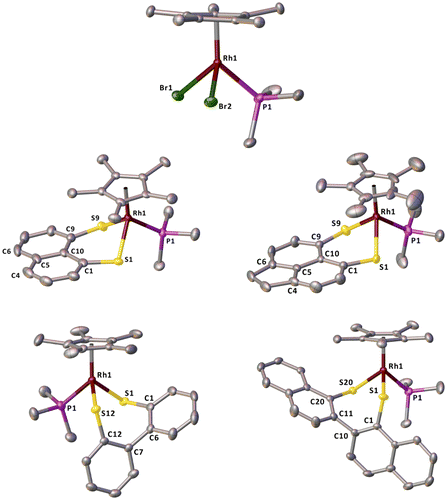
Figure 5. Crystal structures of 1 (Top), 2a (Middle left), 2b (Middle right), 2c (Bottom left) and 2d (Bottom right). Hydrogen atoms are omitted from all structures for clarity. Ellipsoids are plotted at the 50% probability level.
Beyond the electron transfer mimics, there are few examples of complexes incorporating ligand C. Two molybdenum complexes have been reported with one containing an Mo oxygen triple bond (Conry & Tipton, Citation2001; McNaughton, Tipton, Rubie, Conry, & Kirk, Citation2000). In addition, two methods to synthesise titanocene-2,2’-dithiolatobiphenyl have been published (Aucott et al., Citation2005; Stafford, Rauchfuss, Verma, & Wilson, Citation1996). The 2,2’-binaphthalene-based ligand, D, has been largely overlooked with regards to its complexation chemistry compared to the 1,1’-binaphthalene derivative. The only work reported involving this ligand thus far has been its preparation by Armarego (Citation1960), the single-crystal X-ray structure (Kempe, Sieler, Hintzsche, & Schroth, Citation1993), one titanium complex (Aucott et al., Citation2005) and a platinum complex (Aucott, Kilian, Robertson, Slawin, & Woollins, Citation2006).
We have recently published two papers investigating the use of ligands A-C in the formation of half sandwich rhodium and iridium complexes (Nejman, Morton-Fernandez, Black, et al., Citation2015; Nejman, Morton-Fernandez, Moulding, et al., Citation2015). This paper describes the synthesis of a further four half sandwich rhodium (III) dithiolato complexes bearing a neutral phosphine donor. In this contribution, a different phosphine donor has been used compared to the previous work (trimethylphosphine instead of triethylphosphine) (Nejman, Morton-Fernandez, Black, et al., Citation2015). Additionally, a new ligand has been investigated [2,2’-binaphthalene]-1,1’-dithiolate (D). Two synthetic methods were employed due to the varying difficulty in preparing the ligand precursors. Both of these methods were different to the procedures used within our previous work (Nejman, Morton-Fernandez, Black, et al., Citation2015; Nejman, Morton-Fernandez, Moulding, et al., Citation2015). Furthermore, the single-crystal X-ray structure of the complex precursor, [Cp*Rh(PMe3)Br2] (1), is reported for the first time. All the complexes have been fully characterised, principally by multinuclear NMR spectroscopy, mass spectrometry and single-crystal X-ray diffraction.
2. Results and discussion
2.1. Synthetic methods
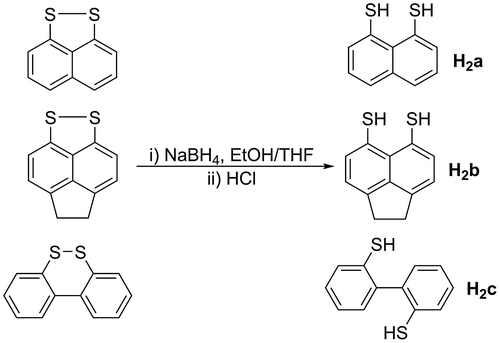
The dithiol pro-ligands [Naphth(SH)2] (H2a), [Acenap(SH)2] (H2b) and [Biphen(SH)2] (H2c) were prepared from their respective disulphides, naphtho[1,8-cd]-1,2-dithiole (Ashe, Kampf, & Savla, Citation1994), 5,6-dihydroacenaphtho[5,6-cd]-1,2-dithiole (Benson et al., Citation2013) and dibenzo[c,e]-1,2-dithiine (Cossu, Delogu, Fabbri, & Maglioli, Citation1991). The reduction of the disulphides was performed using NaBH4 followed by an acidic work up which afforded the three pro-ligands (Figure ) (Yui, Aso, Otsubo, & Ogura, Citation1988). The disulphide precursor to D, [2,2’-BinapS2], was prepared according to the literature procedure by Armarego (Citation1960). The reduction to the dithiol was not attempted as the amount of disulphide prepared was not sufficient. For this reason, the reduction to the reactive dithiolate was performed in situ using lithium triethylborohydride.
Figure 6. Synthesis of the pro-ligands H2a-c.
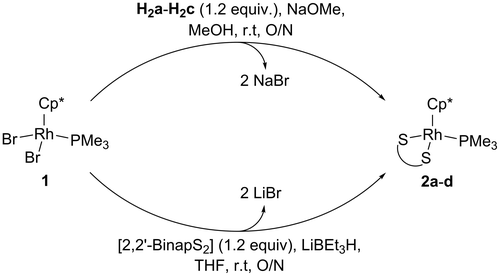
The synthesis of [Cp*Rh(PMe3)(NaphthS2)] (2a), [Cp*Rh(PMe3)(AcenapS2)] (2b), [Cp*Rh(PMe3)(BiphenS2)] (2c) and [Cp*Rh(PMe3)(2,2’-BinapS2)] (2d) is shown in Figure . The metathesis of the bromide ligands with dithiolates A-D proceeds smoothly at room temperature. The presence of the phosphine group allowed the reaction progress to be monitored by 31P NMR spectroscopy. In the case of 2a-c, this meant the loss of the signal from 1, whilst for 2d, the product and 1 are soluble in THF so the conversion could be observed. Upon completion of the reaction for 2a-c filtration of the precipitate followed by washing with methanol was sufficient to afford pure compound. For 2d, purification by column chromatography was required (silica/CH2Cl2). Excellent isolated yields of between 82 and 91% were obtained after purification. Compared to the similar complexes incorporating triethylphosphine, this represents an increase in the isolated yield of approximately 10% (Nejman, Morton-Fernandez, Black, et al., Citation2015). The synthesis of 2a-d all proceeded via a reactive dithiolate intermediate, whereas the triethylphosphine derivatives were prepared by reacting H2a-c directly with the dichloro rhodium precursor. The dithiols represent a less reactive sulfur centre which could explain the higher yields obtained for the complexes reported here.
Figure 7. Reaction conditions for the preparation of 2a-d.
2.2. Data analysis
The 1H NMR spectra (CDCl3) for 2a and 2b show the expected signals, with splitting, from the aromatic backbones in the range of 7.88–6.91 ppm. In the case of 2c and 2d, we observe 8 and 10 signals, respectively, as the two joined aryl ring systems are inequivalent. This is due to the inability of the ligand backbones to rotate around the aryl–aryl bond. Four of the aromatic signals observed for 2c appear as a pseudo triplet of doublets instead of the expected doublet of doublet of doublets. This is due to the 3JHH coupling constants observed between Hb and Ha as well as Hb and Hc (Figure ) being almost identical. Two of these signals overlap closely (δH 7.19 and 7.18 ppm, Figure ); however, both can be distinctly observed and the coupling constants easily extracted. These observations mirror those made for other similar rhodium complexes we have prepared incorporating ligands A-C (Nejman, Morton-Fernandez, Black, et al., Citation2015). For 2d, this signal is not observed as the equivalent positions from each of the two naphthalene ring systems overlap resulting in multiplets. The η5–Cp* methyl signals range from 1.58 to 1.42 ppm and are split into doublets by long range phosphorus coupling (4JHp = 3.0–3.2 Hz). The signals from the methyl groups attached to the phosphorus atom appear as a doublet of doublets for 2a-c, with 3JHP coupling (3JHp = 10.3–10.5 Hz) and long range 4JHRh coupling (4JHRh = 0.6–0.7 Hz). Only a doublet is observed for this signal in 2d with a similar 3JHP coupling to that seen in complexes 2a-c.
The 31P{1H} NMR spectra (CDCl3) for 1 and 2a-d are shown in Table . Complexes 2a-d all display an upfield shift in the 31P{1H} NMR spectra compared to the starting material 1 (Δδ = 0.7–3.3 ppm). The coordination of the dithiolate ligand is also accompanied by a small increase in the 1JPRh coupling (Δ1JPRh = 11–15 Hz) in 2a-d when compared to 1. Both of these observations match those made in the 31P{1H} NMR spectra of the triethylphosphine derivatives of 2a-c (Nejman, Morton-Fernandez, Black, et al., Citation2015).
Table 1. 31P{1H} NMR data (CDCl3) for 1 and 2a-d. All δ values are in ppm and J values are in hertz
As expected, the 13C{1H} NMR spectra (CDCl3) of 2a-d mirrors that of the 1H NMR spectra, with distinct signals for all carbons in the biphenyl and binaphthyl examples. Interestingly, only one of the quaternary carbons bound to the sulfur atoms is split into a doublet (3JCp = 6.5 Hz (2c) and 6.7 Hz (2d)) by the phosphorus atom in 2c and 2d. This provides further support of the difference between the two sides of the aryl–aryl bond. The mass spectra of 2a-d each showed a peak corresponding to [M–PMe3 + H]+ ions at m/z 429, 455, 455 and 555, respectively. Homogeneity of the complexes 2a-d was confirmed by means of accurate elemental analysis.
2.3. Single-crystal X-ray diffraction
Despite the previously reported synthesis and spectroscopic characterisation of precursor complex 1 (Jones & Feher, Citation1984), its single-crystal X-ray data have not been published. Therefore, for completeness we include it here. The crystal structures of 1 and 2a-d are shown below in Figure with selected structural parameters in Tables and .
Table 2. Selected bond lengths [Å] and angles [°] for 1
Table 3. Selected bond lengths [Å], angles [°] and displacements [Å] for 2a-d
All of the complexes, 1 and 2a-d, adopt the piano stool geometry around the rhodium centre we have seen previously (Nejman, Morton-Fernandez, Black, et al., Citation2015; Nejman, Morton-Fernandez, Moulding, et al., Citation2015). The η5–Cp* ring in 1 is slightly tilted as the Rh–C bond lengths vary from 2.141(4) to 2.252(4) Å. The Rh–Br bond lengths (2.550(1) and 2.529(1) Å) and Rh–P bond length (2.284(2) Å) are similar to those previously reported within the Cambridge Structural Database for compounds of a similar type (Rh–Br; 2.543 Å, Rh–P; 2.288 Å) (Bruno et al., Citation2002; Macrae et al., Citation2008; Thomas et al., Citation2010). The angles around the rhodium centre vary with the two P–Rh–Br angles being below the idealised 90°. This is accompanied by a widening of the Br–Rh–Br angle to 94.82(2)° as the two larger atoms try and sit further apart.
The Rh–S bond lengths of 2a and 2b were almost identical (2a; 2.331(1) and 2.332(1) Å, 2b; 2.330(2) Å), whilst in 2c and 2d there was more variation and they were slightly longer (2c; 2.3691(8) and 2.3705(8) Å, 2d; 2.3677(7) and 2.3994(8) Å). These are comparable to other half sandwich complexes with Rh–S bonds reported by ourselves and Jin and co-workers ranging from 2.340 to 2.386 Å (Nejman, Morton-Fernandez, Black, et al., Citation2015; Wang, Lin, Blacque, Berke, & Jin, Citation2008; Xiao & Jin, Citation2008; Yao, Xu, Huo, & Jin, Citation2013). The Rh–P bond lengths show no appreciable change compared to 1 with little variation across the series of 2a-d.
All of the non-Cp* angles around the rhodium centre are reduced to less than 90° for 2a and 2b. This is a consequence of the rigid ligand backbone preventing the sulfur atoms from adopting a more ideal geometry. The effect is most obvious for the naphthalene system as the peri positions are restricted to a slightly shorter distance than those in the acenaphthene system. For both 2a and 2b, the splay angles are large and positive (2a; 19.2(5)°, 2b; 21.0(3)°) as the rhodium centre forces the sulfur atoms apart. The S1–C1···C9–S9 torsion angle is larger in 2a than 2b, again as a consequence of the more limited movement of the sulfur atoms imposed by the backbone. The central C–C–C–C torsion angles are similar in both complexes showing limited buckling of the ring system. The out of plane displacement of the sulfur atoms is slightly greater in 2a than 2b.
In complexes 2c and 2d the non-Cp* angles show a broader range than in 2a and 2b ranging from 88.79(3)–93.94(3)° to 84.11(3)–96.29(3)°, respectively. The ability of the backbone to twist around the aryl–aryl bond allows the sulfur atoms to adopt a more idealised geometry. The torsion angle between the two aryl rings is larger in 2d (79.0(4)°) than 2c (68.0(4)°) most likely due to the added steric bulk of having a binaphthyl instead of biphenyl-based system. In both 2c and 2d, the out of plane displacement of the sulfur atoms are similar.
3. Conclusions
We have prepared and fully characterised a series of new rhodium(III) η5–e have prepared and fully characterised a series of new rhodium(III) ηad 3)Br2] with a series of dithiolates attached to aromatic backbones. Similar features were seen in both the NMR spectra and single-crystal X-ray structures compared to previous rhodium complexes incorporating ligands A-C we have reported (Nejman, Morton-Fernandez, Black, et al., Citation2015; Nejman, Morton-Fernandez, Moulding, et al., Citation2015). The work herein clearly demonstrates the utility of these sulfur ligands in organometallic complexes, with complexes of this type having potential uses in the formation of multimetallic systems.
4. Experimental
4.1. General
Unless otherwise stated all manipulations were performed under an oxygen-free nitrogen atmosphere using standard Schlenk techniques and glassware. Solvents were collected from an MBraun Solvent Purification System or dried and stored according to common procedures (Armarego & Chai, Citation2009). [Cp*Rh(PMe3)Br2] was prepared following literature procedures (Ojima, Vu, & Bonafoux, Citation2002). The disulphide ligand precursors were made according to literature methods (Armarego, Citation1960; Ashe et al., Citation1994; Benson et al., Citation2013; Cossu et al., Citation1991). The pro-ligand H2a was prepared following the literature procedure (Yui et al., Citation1988), with H2c prepared following an identical procedure. H2b was prepared according to literature (Nejman, Morton-Fernandez, Black, et al., Citation2015). 1H, 13C{1H}, 31P and 31P{1H} NMR spectra were obtained on either a Bruker Avance II 400 or Bruker Avance III 500 spectrometer. Full assignments of the 1H and 13C{1H} NMR spectra were made with the aid of H–H DQF COSY, H–C HSQC and H–C HMBC experiments. For 1H and 13C{1H} spectra δH and δC are reported relative to TMS, residual solvent peaks (CDCl3; δH 7.26, δC 77.2 ppm) were used for calibration. For 31P and 31P{1H} spectra δP are reported relative to external 85% H3PO4. All measurements were performed at 21°C with shifts reported in ppm. p-td has been used to denote a pseudo-triplet of doublet. IR spectra were collected on a Perkin Elmer 2000 NIR/Raman Fourier transform spectrometer with a dipole pumped NdYAG near-IR excitation laser. Mass spectra were acquired by the EPSRC UK National Mass Spectrometry Facility at Swansea University. Elemental analysis was performed by Stephen Boyer at the London Metropolitan University.
4.2. Dithiolato complexes
4.2.1. [Cp*Rh(PMe3)(NaphthS2)] (2a)
A methanol (20 mL) solution of [Cp*Rh(PMe3)Br2] (120 mg, 0.25 mmol), [Naphth(SH)2] (60 mg, 0.31 mmol) and NaOMe (17 mg, 0.31 mmol) was stirred at room temperature overnight. The red precipitate was filtered, washed with MeOH then dried under vacuum for 3 h. The product was obtained as a red solid (110 mg, 0.21 mmol, 83%). Crystals suitable for X-ray work were obtained by slow evaporation from CH2Cl2. Anal. calcd. for C23H30PRhS2 (504.06 g mol−1): C, 54.76; H, 5.99. Found: C, 54.69; H, 5.96. 1H NMR (400 MHz, CDCl3): δ 7.88 (dd, 3JHH = 7.3, 4JHH = 1.3 Hz, 2 H, H2,8), 7.47 (dd, 3JHH = 8.1, 4JHH = 1.1 Hz, 2 H, H4,6), 7.05 (dd, 3JHH = 8.1 & 7.3 Hz, 2 H, H3,7), 1.54 (dd, 2JHp = 10.3, 3JHRh = 0.7 Hz, 9 H, PMe3), 1.49 (d, 4JHp = 3.0 Hz, 15 H, Cp*–Me). 13C{1H} NMR (100 MHz, CDCl3): δ 139.5 (d, 3JCp = 5.4 Hz, Cq, C1,9), 136.3 (Cq, C5), 133.9 (Cq, C10), 128.1 (CH, C2,8), 124.9 (CH, C4,6), 123.8 (CH, C3,7), 99.7 (dd, 1JCRh = 4.5, 2JCp = 2.9 Hz, Cq, Cp*), 14.9 (d, 1JCp = 32.7 Hz, CH3, PMe3), 8.9 (CH3, Cp*). 31P NMR (162 MHz, CDCl3): δ 1.7 (br d, 1JPRh = 149 Hz). 31P{1H} NMR (162 MHz, CDCl3): δ 1.7 (d, 1JPRh = 150 Hz). HRMS (APCI+): m/z (%) Calcd. for C20H22RhS2: 429.0212, found 429.0209 (100) [M–PMe3 + H]. IR (KBr): νmax/cm−1 3040w (νAr-H), 2907 m (νC-H), 1536s, 1195 m, 952s, 810 m, 761 m.
4.2.2. [Cp*Rh(PMe3)(AcenapS2)] (2b)
A methanol (20 mL) solution of [Cp*Rh(PMe3)Br2] (120 mg, 0.25 mmol), [Acenap(SH)2] (68 mg, 0.31 mmol) and NaOMe (17 mg, 0.31 mmol) was stirred at room temperature overnight. The red precipitate was filtered, washed with MeOH then dried under vacuum for 3 h. The product was obtained as a red solid (121 mg, 0.22 mmol, 91%). Crystals suitable for X-ray work were obtained by slow evaporation from CH2Cl2. Anal. calcd. for C25H32PRhS2 (530.07 g mol−1): C, 56.60; H, 6.08. Found: C, 56.49; H, 6.11. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 3JHH = 7.2 Hz, 2 H, H2,8), 6.91 (d, 3JHH = 7.2 Hz, 2 H, H3,7), 3.17 (s, 4 H, H11,12), 1.54 (dd, 2JHp = 10.3, 3JHRh = 0.7 Hz, 9 H, PMe3), 1.50 (d, 4JHp = 3.0 Hz, 15 H, Cp*–Me). 13C{1H} NMR (100 MHz, CDCl3): δ 141.7 (Cq, C4,6), 141.1 (Cq, C5), 134.8 (d, 2JCRh = 5.5 Hz, Cq, C1,9), 132.3 (Cq, C10), 128.6 (CH, C2,8), 117.9 (CH, C3,7), 99.5 (dd, 1JCRh = 4.9, 2JCp = 2.9 Hz, Cq, Cp*), 30.1 (CH2, C11,12), 15.0 (d, 1JCp = 33.0 Hz, CH3, PMe3), 8.9 (CH3, Cp*). 31P NMR (162 MHz, CDCl3): δ 2.9 (br d, 1JPRh = 148 Hz). 31P{1H} NMR (162 MHz, CDCl3): δ 2.9 (d, 1JPRh = 148 Hz). HRMS (APCI+): m/z (%) Calcd. for C22H24RhS2: 455.0369, found 455.0362 (80) [M–PMe3 + H], 216.0060 (95) [C12H8S2], 184.0339 (55) [C12H8S], 152.0618 (100) [C12H8]. IR (KBr): νmax/cm−1 3037w (νAr-H), 2907 m (νC–H), 1552 m, 1404 m, 1027 m, 952s, 837 m, 734 m.
4.2.3. [Cp*Rh(PMe3)(BiphenS2)] (2c)
A methanol (20 mL) solution of [Cp*Rh(PMe3)Br2] (120 mg, 0.25 mmol), [Biphen(SH)2] (71 mg, 0.33 mmol) and NaOMe (19 mg, 0.33 mmol) was stirred at room temperature overnight. The red precipitate was filtered, washed with MeOH then dried under vacuum for 3 h. The product was obtained as a red solid (110 mg, 0.21 mmol, 83%). Crystals suitable for X-ray work were obtained by slow evaporation from CH2Cl2. Anal. calcd. for C25H32PRhS2 (530.07 g mol−1): C, 56.60; H, 6.08. Found: C, 56.49; H, 6.15. 1H NMR (400 MHz, CDCl3): δ 7.66 (dd, 3JHH = 7.6, 4JHH = 1.3 Hz, 1 H, H2), 7.64 (dd, 3JHH = 7.7, 4JHH = 1.3, 1 H, H11), 7.19 (p-td, 3JHH = 7.5, 4JHH = 1.4 Hz, 1 H, H4), 7.18 (p-td, 3JHH = 7.6, 4JHH = 1.4 Hz, 1 H, H9), 7.03 (p-td, 3JHH = 7.6, 4JHH = 1.6 Hz, 1 H, H3), 6.98 (p-td, 3JHH = 7.6, 4JHH = 1.6 Hz, 1 H, H10), 6.94 (dd, 3JHH = 7.5, 4JHH = 1.5 Hz, 1 H, H5), 6.86 (dd, 3JHH = 7.5, 4JHH = 1.5 Hz, 1 H, H8), 1.58 (d, 4JHp = 3.2 Hz, 15 H, Cp*–Me), 1.39 (dd, 2JHp = 10.5, 3JHRh = 0.6 Hz, 9 H, PMe3). 13C{1H} NMR (100 MHz, CDCl3): δ 151.0 (Cq, C6), 150.0 (Cq, C7), 143.0 (d, 3JCp = 6.5 Hz, Cq, C1), 140.2 (Cq, C12), 137.2 (CH, C2), 135.2 (CH, C11), 130.9 (CH, C8), 130.6 (CH, C5), 126.2 (CH, C3,9), 125.7 (CH, C4), 125.5 (CH, C10), 99.3 (dd, 1JCRh = 5.3, 2JCp = 3.2 Hz, Cq, Cp*), 15.9 (d, 1JCp = 31.6 Hz, CH3, PMe3), 8.8 (CH3, Cp*). 31P NMR (162 MHz, CDCl3): δ 0.3 (br d, 1JPRh = 153 Hz). 31P{1H} NMR (162 MHz, CDCl3): δ 0.3 (d, 1JPRh = 152 Hz). MS (APCI+): m/z (%) Calcd. for C22H24RhS2: 455.0369, found 455.0365 (100) [M–PMe3 + H]. IR (KBr): νmax/cm−1 3037w (νAr-H), 2905 m (νC-H), 1451 m, 1404 m, 1280w, 959s, 750s.
4.2.4. [Cp*Rh(PMe3)(2,2’-BinapS2)] (2d)
To a THF (15 mL) solution of [2,2’-BinapS2] (117 mg, 0.37 mmol) was added lithium triethylborohydride (0.80 mL, 0.80 mmol, 1 M soln in THF) at room temperature. The reaction was left to stir for 0.5 h during which time the solution turned from yellow to almost colourless. To this was added [Cp*Rh(PMe3)Br2] (148 mg, 0.31 mmol) and the solution turned instantly to a very dark red/purple colour. The reaction was left to stir at room temperature overnight. The solvent was removed under vacuum and the crude product purified by column chromatography (silica/CH2Cl2). The solvent was removed to afford the product as a dark purple solid (147 mg, 0.23 mmol, 82%). Crystals suitable for X-ray work were obtained by slow evaporation from CH2Cl2. Anal. calcd. for C33H36PRhS2 (630.11 g mol−1): C, 62.84; H, 5.75. Found: C, 62.73; H, 5.66. 1H NMR (500 MHz, CDCl3): δ 9.20 (d, 3JHH = 8.6 Hz, 1 H, H3), 9.17 (d, 3JHH = 8.5 Hz, 1 H, H18), 7.80 (d, 3JHH = 8.3 Hz, 1 H, H6), 7.77 (d, 3JHH = 8.2 Hz, 1 H, H15), 7.65 (d, 3JHH = 8.3 Hz, 1 H, H13), 7.64 (d, 3JHH = 8.2 Hz, 1 H, H8), 7.54–7.48 (m, 2 H, H4,17), 7.45–7.39 (m, 2 H, H5,16), 7.01 (d, 3JHH = 8.3 Hz, 1 H, H9), 6.86 (d, 3JHH = 8.3 Hz, 1 H, H12), 1.42 (d, 4JHp = 3.2 Hz, 15 H, Cp*–Me3), 1.31 (d, 2JHp = 10.6 Hz, 9 H, PMe3). 13C{1H} NMR (125 MHz, CDCl3): δ 150.1 (Cq, C11), 148.1 (Cq, C10), 140.8 (d, 3JCp = 6.7 Hz, Cq, C1), 138.3 (Cq, C2), 138.0 (Cq, C19), 137.5 (Cq, C20), 132.9 (Cq, C14), 132.1 (Cq, C7), 129.5 (CH, C12), 129.2 (CH, C3,9), 128.0 (CH, C15), 127.9 (CH, C18), 127.7 (CH, C6), 125.4 (CH, C17), 125.0 (CH, C13), 124.8 (CH, C8,16), 124.7 (CH, C5), 124.3 (CH, C4), 99.7 (dd, 1JCRh = 5.5, 2JCp = 3.5 Hz, Cq, Cp*), 15.7 (d, 1JCp = 31.5 Hz, CH3, PMe3), 9.2 (CH3, Cp*). 31P NMR (202 MHz, CDCl3): δ 2.2 (br d, 1JPRh = 152 Hz). 31P{1H} NMR (202 MHz, CDCl3): δ 2.2 (d, 1JPRh = 152 Hz). HRMS (APCI+): m/z (%) Calcd. for C30H28RhS2: 555.0687, found 555.0729 (90) [M–PMe3 + H], 284.0738 (100) [C20H12S]. IR (KBr): νmax/cm−1 3044w (νAr-H), 2906 m (νC-H), 1493 m, 1281 m, 949s, 815s, 747s, 672 m, 546w.
4.3. Crystal structure analysis
Tables and list the details of data collections and refinements. Data for 1, 2b, 2c and 2d were collected using a Rigaku SCX-Mini (Mo–Kα, graphite monochromator) at −100°C; for 2b using a Rigaku Saturn724 at −148°C. Intensities were corrected for Lorentz polarisation, and adsorption. Structures were solved by direct methods and refined by full-matrix least-squares against F2 (SHELXL) (Sheldrick, Citation2008). Hydrogen atoms were assigned riding isotropic displacements parameters and constrained to idealised geometries. Non-hydrogen atoms were refined anisotropically. CCDC No. 1489639–1489643.
Table 4. Crystallographic data for complexes 1, 2a and 2b
Table 5. Crystallographic data for complexes 2c and 2d
Additional information
Funding
Notes on contributors
J. Derek Woollins
J. Derek Woollins has a long-standing interest in the synthetic and structural chemistry of group 16 elements and their coordination compounds. Recent work has investigated peri substituted naphthalenes and related systems, and the work described here examines the coordination of some peri and related ligand systems.
Related Research Data
References
- Albers, A., Demeshko, S., Dechert, S., Saouma, C. T., Mayer, J. M., & Meyer, F. (2014). Fast proton-coupled electron transfer observed for a high-fidelity structural and functional [2Fe–2S] Rieske model. Journal of the American Chemical Society, 136, 3946–3954.10.1021/ja412449v
- Armarego, W. L. F. (1960). 83. The synthesis of two dinaphthothiophens. Journal of the Chemical Society (Resumed), 1960 433–436.10.1039/jr9600000433
- Armarego, W. L. F., & Chai, C. L. L. (2009). Purification of Laboratory Chemicals (6th ed.). Oxford: Butterworth-Heinemann.
- Ashe, A. J., Kampf, J. W., & Savla, P. M. (1994). The reaction of sulfur with dilithio compounds. The syntheses and structures of phenanthro [1, 10-cd]-1,2-dithiole and phenanthro[4,5-cde] [1,2]dithiin. Heteroatom Chemistry, 5, 113–119.10.1002/(ISSN)1098-1071
- Aucott, S. M., Kilian, P., Milton, H. L., Robertson, S. D., Slawin, A. M. Z., & Woollins, J. D. (2005). Bis(Cyclopentadienyl)Titanium Complexes of Naphthalene−1,8-Dithiolates, Biphenyl 2,2‘-Dithiolates, and Related Ligands. Inorganic Chemistry, 44, 2710–2718.10.1021/ic048483l
- Aucott, S. M., Kilian, P., Robertson, S. D., Slawin, A. M. Z., & Woollins, J. D. (2006). Platinum complexes of dibenzo[1,2]dithiin, dibenzo[1,2]dithiin oxides and related polyaromatic hydrocarbon ligands. Chemistry - A European Journal, 12, 895–902.10.1002/(ISSN)1521-3765
- Ballmann, J., Dechert, S., Demeshko, S., & Meyer, F. (2009). Tuning electronic properties of biomimetic [2Fe-2S] clusters by ligand variations. European Journal of Inorganic Chemistry, 3219–3225.10.1002/ejic.v2009:22
- Benson, C. G. M., Schofield, C. M., Randall, R. A. M., Wakefield, L., Knight, F. R., Slawin, A. M. Z., & Woollins, J. D. (2013). Platinum Complexes of 5,6-Dihydroacenaphtho[5,6-cd]-1,2-dichalcogenoles. European Journal of Inorganic Chemistry, 427–437.10.1002/ejic.201200967
- Bond, A. M., & Martin, R. L. (1984). Electrochemistry and redox behaviour of transition metal dithiocarbamates Coordination chemistry reviews, 54, 23–98.10.1016/0010-8545(84)85017-1
- Bosman, W. P., & van der Linden, H. G. M. (1977). Tris[triphenylphosphinehexachloronaphthalene-1,8-dithiolatonickel(II)], [Ni3(PPh3)3(S2C10Cl6)3], a Ni3S6 metal–sulphur cluster compound; preparation and crystal and molecular structure. Journal of the Chemical Society, Chemical Communications, 714–715.10.1039/C39770000714
- Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J., & Taylor, R. (2002). New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallographica Section B Structural Science, 58, 389–397.10.1107/S0108768102003324
- Burns, R. P., & McAuliffe, C. A. (1979). 1,2-Dithiolene complexes of transition metals. Advances in Inorganic Chemistry and Radiochemistry, 22, 303–348.
- Burns, R. P., McCullough, F. P., & McAuliffe, C. A. (1980). 1,1-Dithiolato complexes of the transition elements. Advances in Inorganic Chemistry and Radiochemistry, 23, 211–280.
- Charreteur, K., Kdider, M., Capon, J.-F., Gloaguen, F., Pétillon, F. Y., Schollhammer, P., & Talarmin, J. (2010). Effect of electron-withdrawing dithiolate bridge on the electron-transfer steps in diiron molecules related to [2Fe](H) subsite of the [FeFe]-hydrogenases. Inorganic Chemistry, 49, 2496–2501.10.1021/ic902401k
- Conry, R., & Tipton, A. A. (2001). A mononuclear molybdenum(V) mono-oxo biphenyl-2,2′-dithiolate complex in which the metal resides within a cleft formed by the ligands and that exhibits N-H ... S hydrogen bonding in the solid state. JBIC Journal of Biological Inorganic Chemistry, 6, 359–366.10.1007/s007750100207
- Cossu, S., Delogu, G., Fabbri, D., & Maglioli, P. (1991). A rapid preparation of 2,2′-dimercaptobiphenyl. Organic Preparations and Procedures International, 23, 455–457.10.1080/00304949109458237
- Eisenberg, R. (2007). Structural systematics of 1,1- and 1,2-dithiolato chelates. In Progress in Inorganic Chemistry (pp. 295–369). John Wiley & Sons.
- Figliola, C., Male, L., Horton, P. N., Pitak, M. B., Coles, S. J., Horswell, S. L., & Grainger, R. S. (2014). [FeFe]-Hydrogenase synthetic mimics based on peri-substituted dichalcogenides. Organometallics, 33, 4449–4460.10.1021/om500683p
- Figliola, C., Male, L., Horswell, S. L., & Grainger, R. S. (2015). N-Derivatives of peri-Substituted Dichalcogenide [FeFe]-Hydrogenase Mimics: Towards Photocatalytic Dyads for Hydrogen Production. European Journal of Inorganic Chemistry, 2015, 3146–3156.10.1002/ejic.v2015.19
- Jones, W. D., & Feher, F. J. (1984). Preparation and conformational dynamics of (C5Me5)Rh(PR13)RX. Hindered rotation about rhodium-phosphorus and rhodium-carbon bonds. Inorganic Chemistry, 23, 2376–2388.10.1021/ic00184a005
- Kempe, R., Sieler, J., Hintzsche, E., & Schroth, W. (1993). Crystal structure of dinaphto[l,2-c][2′,1′-e]-1,2-dithiin, C20H12S2. Zeitschrift für Kristallographie, 207, 314–315.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., … Wood, P. A. (2008). Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. Journal of Applied Crystallography, 41, 466–470.10.1107/S0021889807067908
- McNaughton, R. L., Tipton, A. A., Rubie, N. D., Conry, R. R., & Kirk, M. L. (2000). Electronic structure studies of oxomolybdenum tetrathiolate complexes: Origin of reduction potential differences and relationship to cysteine−molybdenum bonding in sulfite oxidase. Inorganic Chemistry, 39, 5697–5706.10.1021/ic0003729
- Nejman, P. S., Morton-Fernandez, B., Black, N., Cordes, D. B., Slawin, A. M. Z., Kilian, P., & Woollins, J. D. (2015). The preparation and characterisation of rhodium(III) and Iridium(III) half sandwich complexes with napthalene-1,8-dithiolate, acenaphthene-5,6-dithiolate and biphenyl-2,2′-dithiolate. Journal of Organometallic Chemistry, 776, 7–16.10.1016/j.jorganchem.2014.10.023
- Nejman, P. S., Morton-Fernandez, B., Moulding, D. J., Athukorala Arachchige, K. S., Cordes, D. B., Slawin, A. M. Z., … Woollins, J. D. (2015). Structural diversity of bimetallic rhodium and iridium half sandwich dithiolato complexes. Dalton Transactions, 44, 16758–16766.10.1039/C5DT02542G
- Ojima, I., Vu, A. T., & Bonafoux, D. (2002). Product Class 5: Organometallic complexes of rhodium. Science of Synthesis, 1, 531–616.
- Phillips, J. R., Poat, J. C., Slawin, A. M. Z., Williams, D. J., Wood, P. T., & Woollins, J. D. (1995). Polymeric and bimetallic complexes of diisopropyl monothiophosphate. Journal of the Chemical Society, Dalton Transactions, 2369–2375.10.1039/dt9950002369
- Samuel, A. P. S., Co, D. T., Stern, C. L., & Wasielewski, M. R. (2010). Ultrafast photodriven intramolecular electron transfer from a zinc porphyrin to a readily reduced diiron hydrogenase model complex. Journal of the American Chemical Society, 132, 8813–8815.10.1021/ja100016v
- Sheldrick, G. (2008). Acta Crystallographica Section A Foundations of Crystallography. International Union of Crystallography, International Union of Crystallography, 64, 112–122.10.1107/S0108767307043930
- Stafford, P. R., Rauchfuss, T. B., Verma, A. K., & Wilson, S. R. (1996). Titanocene complexes of ring-opened dibenzothiophene and related dimercaptobiaryl ligands. Journal of Organometallic Chemistry, 526, 203–214.10.1016/S0022-328X(96)06581-3
- Teo, B. K., & Snyder-Robinson, P. A. (1978). Metal tetrathiolenes. 3. Structural study of the first bimetallic tetrathiolene complex, tetrakis(triphenylphosphine)diplatinum tetrathionapthalene, (Ph3P)4Pt2(C10H4S4) revealing an unexpected molecular distortion. Inorganic Chemistry, 17, 3489–3497.10.1021/ic50190a034
- Teo, B. K., & Snyder-Robinson, P. A. (1979a). Metal–tetrathiolene chemistry: the first disulphur bridge in the novel di-iridium cluster (Ph3P)2(CO)2Br2Ir2(C10H4S4). X-Ray crystal structure. Journal of the Chemical Society, Chemical Communications, 255–256.10.1039/C39790000255
- Teo, B. K., & Snyder-Robinson, P. A. (1979b). Metal tetrathiolenes. 4. Synthesis, characterization, and electrochemistry of new discrete diplatinum-tetrathiolene complexes. Inorganic Chemistry, 18, 1490–1495.10.1021/ic50196a017
- Teo, B. K., & Snyder-Robinson, P. A. (1981). Molecular systematics in metal tetrathiolenes. A novel 2-, 4-, 6-, 8-, and 12-electron-donating ligand system upon coordination to one, two, two, three and four metals, respectively. Inorganic Chemistry, 20, 4235–4239.10.1021/ic50226a041
- Teo, B. K., & Snyder-Robinson, P. A. (1984). Metal tetrathiolenes. 8. Molecular structures of two isostructural two-electron systems: (Ph3P)2(CO)XIr(C10Cl4S4) (X-Cl,H). The first member of a novel series of metal tetrathiolene complexes. Inorganic Chemistry, 23, 32–39.10.1021/ic00169a009
- Teo, B. K., Wudl, F., Hauser, J. J., & Kruger, A. (1977). Reactions of tetrathionaphthalene with transition metal carbonyls. Synthesis and characterization of two new organometallic semiconductors (C10H4S4Ni)x and [C10H4S4Co2(CO)2]x and a tetrairon cluster C10H4S4Fe4(CO)12. Journal of the American Chemical Society, 99, 4862–4863.10.1021/ja00456a073
- Teo, B. K., Wudl, F., Marshall, J. H., & Kruger, A. (1977). Synthesis, characterization, and electrochemistry of a new platinum "tetrathiolene" cluster, tetrakis(triphenylphosphine)tetrathionaphthalenediplatinum(II,II): a novel system with five reversible oxidation states. Journal of the American Chemical Society, 99, 2349–2350.10.1021/ja00449a060
- Teo, B. K., Bakirtzis, V., & Snyder-Robinson, P. A. (1983). Syntheses and structures of a novel series of tetrathiolene clusters: (.eta.5-C5H5)2Fe2(C10Cl4S4)(C10Cl4S3), (.eta.5-C5H5)2Co2(C10Cl4S4), and (.eta.5-C5H5)2Ni2(C10Cl4S4). Journal of the American Chemical Society, 105, 6330–6332.10.1021/ja00358a034
- Thomas, I. R., Bruno, I. J., Cole, J. C., Macrae, C. F., Pidcock, E., & Wood, P. A. (2010). WebCSD: the online portal to the Cambridge Structural Database. Journal of Applied Crystallography, 43, 362–366.10.1107/S0021889810000452
- Topf, C., Monkowius, U., & Knör, G. (2012). Design, synthesis and characterization of a modular bridging ligand platform for bio-inspired hydrogen production. Inorganic Chemistry Communications, 21, 147–150.10.1016/j.inoche.2012.04.034
- Tuna, F., Smith, C. A., Bodensteiner, M., Ungur, L., Chibotaru, L. F., McInnes, E. J. L., Winpenny, R. E. P., Collison, D., & Layfield, R. A. (2012). A high anisotropy barrier in a sulfur-bridged organodysprosium single-molecule magnet. Angewandte Chemie International Edition, 51, 6976–6980.10.1002/anie.201202497
- Wang, G.-L., Lin, Y.-J., Blacque, O., Berke, H., & Jin, G.-X. (2008). Helical Supramolecular Assemblies of {2,4,6-[Cp∗Rh(E2-1,2-C2B10H10)(NC5H4CH2S)]3(triazine)} (E = S, Se) Shaped by Cp∗−Toluene−Cp∗ π-Stacking Forces and BH−Pyridine Hydrogen Bonding. Inorganic Chemistry, 47, 2940–2942.10.1021/ic800105h
- Woollins, J. D. (1996). Metallacycles with group 15/16 ligands. In S. C. Mitchell (Ed.), Biological interactions of sulfur compounds (pp. 1–19). London: Taylor & Francis.
- Wright, R. J., Lim, C., & Tilley, T. D. (2009). Diiron proton reduction catalysts possessing electron-rich and electron-poor naphthalene-1,8-dithiolate ligands. Chemistry - A European Journal, 15, 8518–8525.10.1002/chem.v15:34
- Xiao, X.-Q., & Jin, G.-X. (2008). Half-sandwich rhodium complexes containing both N-heterocyclic carbene and ortho-carborane-1,2-dithiolate ligands. Journal of Organometallic Chemistry, 693, 316–320.10.1016/j.jorganchem.2007.11.005
- Yao, Z.-J., Xu, B., Huo, X.-K., & Jin, G.-X. (2013). Homonuclear half-sandwich iridium and rhodium complexes containing dichalcogenolato-functionalized o-carboranyl ligands. Journal of Organometallic Chemistry, 747, 85–89.10.1016/j.jorganchem.2013.03.002
- Yui, K., Aso, Y., Otsubo, T., & Ogura, F. (1988). Syntheses and Properties of 2,2′-Binaphtho[1,8-de]-1,3-dithiinylidene and Its Selenium Analog, 2-(1,3-Dithiol-2-ylidene)naphtho[1,8-de]-1,3-dithiin, and 2-(4H-Thiopyran-4-ylidene)naphtho[1,8-de]-1,3-dithiin. Bulletin of the Chemical Society of Japan, 61, 953–959.10.1246/bcsj.61.953
- Zhou, C.-L., Wang, Z.-M., Wang, B.-W., & Gao, S. (2011). Two Mn6 single-molecule magnets with sulfur-contained capping ligand. Polyhedron, 30, 3279–3283.10.1016/j.poly.2011.05.046