
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nonenzymatic glycation (NEG) begins with the non-covalent binding of a glucopyranose to a protein. The bound glucopyranose must then undergo structural modification to generate a bound electrophile that can reversibly form a Schiff base, which can then lead to Amadori intermediates, and ultimately to glycated proteins. Inorganic phosphate (Pi) is known to accelerate the glycation of human hemoglobin (HbA), although the specific mechanism(s) of Pi as an effector reagent have not been determined. The aim of this study was to determine whether Pi and a glucopyranose can concomitantly bind to HbA and react while bound within the early, noncovalent stages to generate electrophilic species capable of progress in NEG. 31P and 1HNMR of model reactions confirm that bimolecular reactions between Pi and glucopyranose occur generating modified glucose electrophiles. Computations of protein/substrate interactions predict that Pi can concomitantly bind with a glucopyranose in HbA pockets with geometries suitable for multiple acid/base mechanisms that can generate any of four transient electrophiles. Pi-facilitated mechanisms in the noncovalent stages predict that the glycation of β-Val1 of HbA to HbA1c is a “hot spot” because the β-Val1 pocket facilitates many more mechanisms than any other site. The mechanistic diversity of the Pi effect within the early noncovalent stages of NEG predicts well the overall site selectivity observed from the in vivo glycation of HbA in the presence of Pi. These insights extend our basic understanding of the NEG process and may have clinical implications for diabetes mellitus and even normal aging.
Public Interest Statement
Glucose derived from dietary sources or produced by the liver serves as a vital reagent for energy production in cells to support critical cellular functions. Over time, glucose also binds to and reacts with intracellular and extracellular proteins to generate glycated proteins. Elevated blood glucose concentrations and excessive amounts of glycated protein have been linked to complications in patients with diabetes mellitus. Historically, the extent of protein glycation has been linked exclusively to blood glucose concentrations. However, it has been shown that inorganic phosphate (Pi) accelerates the glycation of human adult hemoglobin (HbA) in red blood cells to generate HbA1c. The role of Pi in this process has not been determined. Using computational and experimental techniques, our findings indicate that Pi plays multiple roles in the formation of HbA1c. These results suggest that clinical measures of Pi concentrations in diabetic patients vs. disease progression and in comparisons with normal controls might be insightful in monitoring diabetic complications.
Competing interests
The authors declare no competing interest.
1. Introduction
Glucose is a vital substrate for energy metabolism of all cells, yielding ATP under aerobic and anaerobic conditions. Glucose also reacts nonenzymatically with intracellular and extracellular proteins to form covalently modified proteins (also referred to as glycated proteins), both in vivo (Bunn, Haney, Gabbay, & Gallop, Citation1975; Bunn, Haney, Kamin, Gabbay, & Gallop, Citation1976; Stevens, Vlassara, Abati, & Cerami, Citation1977) and in vitro (Bookchin & Gallop, Citation1968; Lea & Hannan, Citation1949; Mohammad, Fraenkel-Conrat, & Olcott, Citation1949) over prolonged periods of time. This process, termed nonenzymatic glycation (NEG), is a significant feature of diabetes mellitus, in which persistent hyperglycemia leads to the accumulation of advanced glycation endproducts (AGE). NEG is associated with pathophysiological outcomes, as well as age-related chronic diseases such as microangiopathy, retinopathy, and nephropathy (Brownlee, Citation1995).
The most studied NEG reactions are those between glucose and adult human hemoglobin (HbA) yielding HbA1c. HbA1c has a modified glucose bound to the N-terminal valine of one or both of the HbA β-chains and is increased in diabetic patients (Little & Sacks, Citation2009). Measurement of HbA1c has been useful as a clinical marker of long-term (6–8 weeks) glycemic control and is an assessment of the risk of developing cardiovascular disease and diabetic complications.
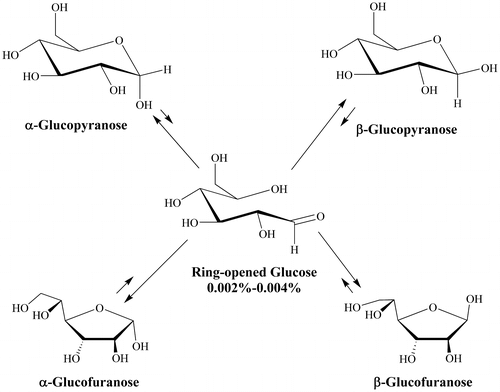
Our research goal was to elucidate the potential roles of inorganic phosphate on the progression of initially bound glucopyranose toward the nonenzymatic glycation of human hemoglobin, with a focus on mechanistic diversity and impacts on site selectivity. More specifically, to better understand what reagents and mechanisms are involved that enable a noncovalently bound glucose isomer to proceed in NEG. Five interconverting glucose isomers exist in reversible equilibrium in the erythrocyte cytosol, including a pair of ring-closed glucopyranoses (α and β; at 34% and 65%, respectively) and a pair of ring-closed glucofuranoses (α and β combined at ca. 1%, Figure ). The ring-closed species are not sufficiently electrophilic (able to accept two electrons in the formation of a new bond) to significantly react with protein amino acid residues and progress in NEG. The fifth structure, through which the four ring-closed isomers interconvert, is a ring-opened isomer containing an aldehyde group. This species is sufficiently electrophilic to react with amino acid residues and progress in NEG. However, the direct binding of the reactive ring-opened isomer of glucose in NEG is unlikely. The equilibrium concentration of the ring-opened glucose in aqueous solution is 0.002% (Bunn & Higgins, Citation1981) or less, corresponding to just 0.12 μM in erythrocyte cytosol. This is likely an overestimate of the reactive concentration of available ring-opened isomer since reversion to a ring-closed isomer occurs rapidly, less than the NMR timescale of 10−4 seconds (Bryant, Citation1983), making it a transient electrophile. It is therefore anticipated that reversion to ring-closed isomers occurs at a rate faster than ring-open isomer binding. In contrast, the ring-closed isomers of glucose are at 50,000 times the concentration, are long-lived, and bind better to HbA than does the ring-opened isomer (Clark, Santin, Bryant, Holman, & Rodnick, Citation2013). Moreover, the initially bound glucose species to human serum albumin (HSA) have been experimentally determined to be ring-closed isomers (Wang et al., Citation2013). This observation was confirmed by Clark et al. (Citation2013) for HbA and is consistent with studies of glucose and other monosaccharides binding to several enzymes (Allen et al., Citation1994; Lee, Bagdasarian, Meng, & Zeikus, Citation1990; Lee & Jeffrey, Citation2005; Thoden, Kim, Raushel, & Holden, Citation2003).
Figure 1. The five interconverting isomers of glucose observed in aqueous media.
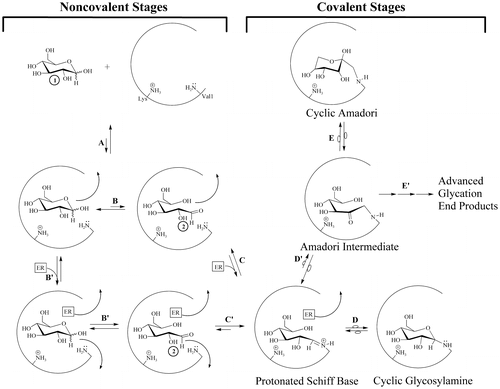
In order to proceed in NEG, the initially bound glucopyranose must undergo modification, while bound, to become sufficiently electrophilic before exiting the protein pocket (for the general scheme of NEG, see Figure ).
Figure 2. The NEG process in the formation of HbA1c involving transient intermediate 2, including routes possible with or without the participation of an effector reagent.
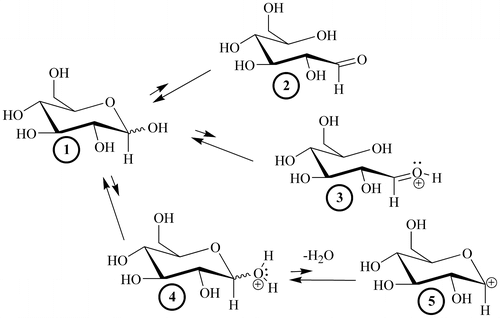
Four potential modified, transient electrophilic glucose species can be reversibly generated from a bound glucopyranose (Structures 2–5, Figure ). Irrespective of the specific structure or formation process, the electrophile can then be attacked by a nucleophilic amino acid residue such as N-terminal valines on the β-chains of HbA and proceed through NEG. For an amino acid residue to act as a nucleophile (donate two electrons in the formation of a new bond with the electrophilic-modified glucose), it must be in an R-NH2 amine form.
Figure 3. The four modified, transient electrophiles (2–5) that can theoretically be generated from a glucopyranose, 1.
Several investigators report that inorganic phosphate (Pi) increases the rate of NEG for certain proteins (Gil, Mata-Segreda, & Schowen, Citation1988; Gil, Peña, Vásquez, & Uzcategui, Citation2002; Kunika, Itakura, & Yamashita, Citation1989; Watkins, Neglia-Fisher, Dyer, Thorpe, & Baynes, Citation1987), although not all proteins (Watkins et al., Citation1987). For HbA, a mechanism(s) for accelerating glycation has not been fully established. That said, most of the mechanisms that have been proposed place the Pi effect (here defined as glycation facilitation by Pi) at Amadori formation or later (Gil, Salcedo, & Romero, Citation2005; Gil, Vásquez, Peña, & Uzcategui, Citation2004; Gil et al., Citation1988, Citation2002; Ito, Nakahari, & Yamamoto, Citation2011; Kunika et al., Citation1989; Watkins et al., Citation1987). Recently, Rodnick, et al. (Citation2017) posited that Pi plays multiple mechanistic roles within the pre-Amadori, early noncovalent stages of HbA glycation. Specifically, Pi is asserted to facilitate the modification of the initially bound glucopyranose to more reactive species.
The first goal of this work was to assess how Pi serves to modify the initially bound glucopyranose to generate a bound transient electrophile. In addition, what transient electrophilic species can theoretically be formed through modification of a bound glucopyranose? Is there mechanistic diversity involving Pi, and if so, how does it impact site selectivity in HbA glycation? Site selectivity was defined here as the relative extent of glycation between HbA pockets and within a given HbA pocket. A clinically relevant objective was to better understand why Val1 of β-chains (HbA1c) predominates over other potential HbA glycation sites and to understand why HbA1c formation occurs so slowly (over weeks to months).
2. Results
1HNMR assessment for the Pi-facilitated formation of reactive electrophiles from a glucopyranose in aqueous media. First, pure α-glucopyranose was placed in aqueous solution (D2O) and its reversible interconversion with the β-glucopyranose isomer was followed as a function of time by 1HNMR at room temperature (Table , Experiment 1). The extent of α-glucopyranose to β-glucopyranose interconversion through the ring-opened glucose isomer (Figure ) was measured and expressed in terms of the time required for inflection to occur. Inflection is here defined as the time required for the formation of a 50:50 mixture of α- and β-glucopyranose isomers from pure α-glucopyranose (Figure , Table ).
Table 1. Time-dependent 1HNMR data for α- to β-glucopyranose inflection through the ring-opened glucose isomer 2 in D2O at ~20°C in the absence or presence of various physiological anions (serving as effector reagents, ER)
Figure 4. 1HNMR of pure α-glucopyranose in D2O at 20°C followed as a function of time.
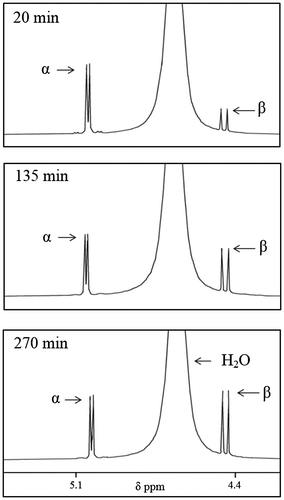
The inflection time in the presence of equimolar dianionic Pi (98% dianionic and 2% monoanionic at pH = 9.1) in D2O is 8 min, an approximate 30-fold decrease relative α- to β-glucopyranose inflection in D2O alone (270 min) (Table , Experiment 2). The inflection time observed with monoanionic Pi (4% dianionic and 96% monoanionic at pH = 4.5) is 215 min, reflecting an enhancement of α- to β-glucopyranose by a factor of 1.25 (Table , Experiment 3). Inflection times for α-G6P to β-G6P are shorter than can be measured (less than 5 min) (Table , Experiment 4).
1HNMR validation that Pi-facilitated formation of reactive electrophiles is the result of Pi structural effect and not a pH effect. The inflection times for six model potential effector reagents (some physiological—histidine, taurine, lactate, bicarbonate, and 2,3-BPG—and one non-physiological—metformin), whose pH values range from 4.2–9.1 (Experiments 5–10) are shown in Table . The reagents were evaluated for mechanistic purposes to discern whether inflection times are governed by effector reagent structure or whether there is a significant pH effect. We observed a large variation in inflection times and inflection times did not trend with pH. Experiments 11–14 were conducted to further assess the facilitation of electrophile formation from α-glucopyranose as affected by Pi mixtures in the physiological pH range (6.4–7.4). Over the physiological pH range, Pi clearly facilitates the formation of electrophiles.
31PNMR and computational validation of a structural effect of Pi: The formation of a glucopyranose: Pi adduct. The 31P chemical shift for Na2HPO4 in D2O (in the absence of glucopyranose) was observed to be 2.78 ppm, while that for NaH2PO4 was 0.47 ppm. When Na2HPO4 was placed in a 1:1 M ratio with α-glucopyranose, a single broadened phosphorous signal at 1.77 ppm was observed. At a 1:5 ratio, the single broadened signal was at 1.66 ppm, while at a 5:1 ratio the signal, again broadened, was at 2.13 ppm. If the Pi were not interacting and making a reversible complex with glucose, the 31P chemical shift should be at 2.78 ppm. That the broadened, single peak observed in a 1:1 dibasic Pi to glucopyranose was at 1.77 ppm in a solution at pH 9.1 makes certain that a new phosphorus environment exists, consistent with a reversible adduct between dibasic Pi and glucopyranose. As computational validation (ab initio HF631G**), energies were calculated for separated α-glucopyranose and Pi and compared to a geometry optimized adduct between α-glucopyranose and Pi. The formation of the bridging adduct was 30 kcal/mol exothermic relative to isolated starting materials. Together, the 31PNMR and ab initio calculations suggest that the primary structural effect of Pi is to bridge with a glucopyranose, enabling the generation of transient electrophiles.
Computational assessment of Pi concomitant binding with glucopyranose isomers in HbA. Binding of dianionic Pi with α-glucopyranose is predicted to take place in three of the four HbA pockets known to be glycated on each β-chain (Table ). These pockets include the β-Val1pocket (containing Val1/Lys8/82/132/144), the Lys17/120 pocket, and the Lys59/61/65/66 pocket. Dianionic Pi is also predicted to concomitantly bind with β–glucopyranose in the β-Val1 pocket. Both α- and β–glucopyranose bind with monoanionic Pi in all four HbA β–chain pockets (including the Lys95 pocket). These concomitant binding events occur with exothermicities comparable with both glucopyranose binding alone (−5.0 to −2.9 kcal/mol) and Pi binding alone [−3.6 to −3.2 kcal/mol, Table ).
Table 2. Computational assessment of α- and β-glucopyranose concomitant binding with Pi to the fully protonated β-chain of oxygenated HbA organized by potential glycation sites, binding exothermicities, and proximate amino acid (AA) residues
Computational assessment of geometrically feasible mechanisms for the Pi-assisted formation of 2–5 from a glucopyranose within the β-Val1 pocket of HbA. Among dozens of energetic minima, six geometries representing six mechanistic classes with the potential to generate one (or more) of 2–5 were found (rendered in a classic arrow-pushing organic formalism for the sake of clarity in Figures –).
Figure 5. Two MOE geometries representative of energetic minima whereby Pi and a glucopyranose concomitantly bind in the β-Val1 pocket of HbA.
For visual clarity, only those residues needed to illustrate the geometrically viable mechanisms are shown. (A) From this geometry, three distinct mechanisms are possible. In one mechanism the dianionic Pi bridges the glucopyranose, protonates the hemiacetal oxygen (dist. = 4.11 Å) and deprotonates the anomeric OH (dist. = 4.23 Å) as Val1 attacks the incipient anomeric carbonyl in a concerted fashion (dist = 5.13 Å) on route to a Schiff base (equating to Figure 5, mechanism I). In a second mechanism, the Lys82 protonates dianionic Pi (dist. = 1.14 Å) as the Pi protonates the anomeric OH (dist. = 4.11 Å), making the anomeric carbon electrophilic, and generating water as a leaving group as the Val1 attacks as a nucleophile in a concerted fashion (dist. = 5.13 Å) on route to a cyclic glycosylamine (equating to Figure 6, Mechanism VIa). In a third mechanism, the Lys82 protonates dianionic Pi (dist. = 1.14 Å) as the Pi protonates the hemiacetal oxygen of the glucopyranose (dist. = 4.11 Å), making the anomeric carbon electrophilic as the Val1 attacks as a nucleophile in a concerted fashion (dist. = 5.13 Å), generating 3 (equating to Figure 6, Mechanism IV) on route to a protonated Schiff base. (B) From this geometry, three distinct mechanisms are possible. In one mechanism the monoanionic Pi bridges the glucopyranose, protonates the hemiacetal oxygen (dist =4.19 Å), and deprotonates the anomeric OH (dist. = 3.60 Å) as Val1 attacks the incipient anomeric carbonyl in a concerted fashion (dist. = 4.91 Å) on route to a Schiff base (equating to Figure 5, mechanism I). In a second mechanism, the monoanionic Pi protonates the anomeric OH (dist. = 1.87 Å), making the anomeric carbon electrophilic, and generating water as a leaving group as the Val1 attacks as a nucleophile in a concerted fashion (dist. = 4.91 Å) on route to a cyclic glycosylamine (equating to Figure 6, Mechanism VIa). In a third mechanism, monoanionic Pi protonates the hemiacetal oxygen of the glucopyranose (dist. = 4.19 Å), making the anomeric carbon electrophilic as the Val1 attacks as a nucleophile in a concerted fashion (dist. = 4.91 Å), generating 3 (equating to Figure 6, Mechanism IV) on route to a Schiff base.
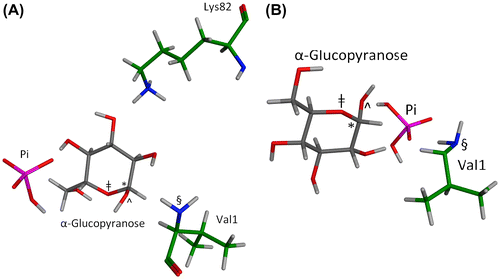
Figure 6. Notes: A two-dimensional rendering of mechanisms for the concomitant binding of Pi (mono- or dianionic) and glucopyranose in HbA as determined by MOE computations.
Pi* refers to the fact that Pi may either dissociate from, or remain within, the pocket as progrees in NEG proceeds.
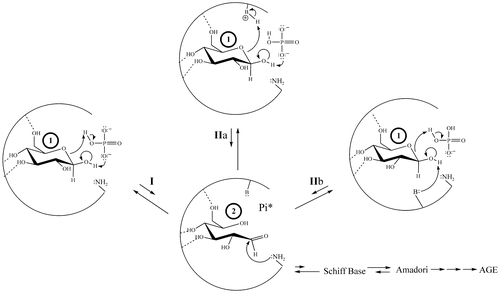
Figure 7. A two-dimensional rendering of the four mechanistic classes for the concomitant binding of Pi and glucopyranose in HbA as determined by MOE computation that go through or generate transient electrophile 3 or 4 (4 may then generate 5).
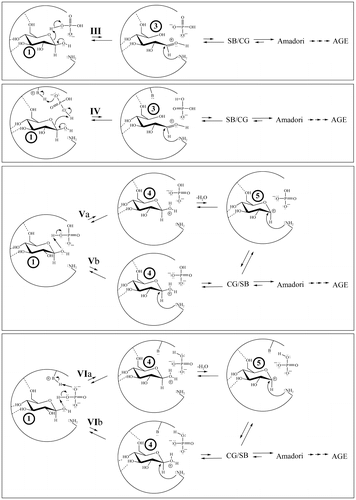
A mechanistic class is defined here as a general reaction process in which the identity of the reagents may vary, but the inherent bond-making/bond-breaking steps are the same.
Computational assessment of geometrically feasible mechanisms for the water-assisted formation of 2–5 from a glucopyranose in HbA. Similar to Pi, a single water molecule can concomitantly bind with either α- or β-glucopyranose in the HbA β-Val1 pocket and exists as an energetic minimum (Supplementary Figure 1). The generation of 2–5 via four mechanistic classes is predicted (Supplementary Figure 2). In addition, concomitant binding of water with each of the glucopyranoses takes place in the other three HbA pockets. In the Lys95 pocket, concomitant binding occurs with suitable geometry for reaction involving Lys95. Concomitant binding of water with each of the glucopyranoses occurs as local minima with suitable geometry to react with Lys61 within the Lys59/61/65/66 pocket. In the Lys17/120 pocket, concomitant binding occurs, but no suitable geometry for reaction is predicted.
Computational assessment of geometrically feasible mechanisms of unassisted formation of 2–5 from a glucopyranose within the β-Val1 pocket of HbA. If a glucopyranose binds alone, the only available acids and bases to generate transient electrophiles are nearby protein amino acid residues. Geometric computations show that a Lys R- can act as an acid, while, in a concerted fashion, a neutral histidine can act as the base to generate transient electrophile 2. Once 2 is formed, a nucleophilic amino acid residue (e.g. β-Val1 or Lys if it is an R-NH2 amine) can then attack in a concerted process (Supplementary Figures 3 and 4).
3. Discussion
Thirty years ago, investigators attributed the Pi-enhanced glycation of HbA (the Pi effect) to the covalent stages of the NEG process (Kunika et al., Citation1989; Watkins et al., Citation1987). This is not contested here. On the basis of the current work, additional Pi effects, specifically within the noncovalent stages of NEG (Figure ), are posited. Glucose isomers must undergo modification while bound to a protein to generate the bound electrophiles necessary for efficient progress in NEG (Clark et al., Citation2013; Rodnick et al., Citation2017; Wang et al., Citation2013). A bound glucopyranose (Figure , Structure 1) can theoretically undergo modification to at least four transient electrophilic species. These include the ring-opened neutral glucose isomer (Structure 2), which is formed by protonation of the hemiacetal oxygen with concerted deprotonation of the anomeric OH. Structure 3, a protonated, ring-opened glucose, is formed by protonation of the hemiacetal oxygen by an acid. Structure 4, a protonated, ring-closed glucose, is generated by the protonation of the hydroxyl oxygen on the anomeric carbon. Structure 4 could react further by losing water to produce an oxocarbenium ion (Structure 5). Unlike 2 and 3, transient structures 4 and 5 remain ring-closed species. Each of these transient species (2–5) is more electrophilic than 1, and is likely to be much more reactive in NEG.
Once formed, 2–5 have three potential outlets. First, nucleophilic attack by an R-NH2 amino acid residue (such as β-Val1 or an internal Lys of HbA) can occur, which constitutes progress from the noncovalent to the covalent stages of NEG (Figure ). Nucleophilic attack on either 2 or 3 would directly form a Schiff base, while nucleophilic attack on 4 or 5 would initially generate a cyclic glycosylamine. Note that the Schiff base and the cyclic glycosylamine are in equilibrium. As a second fate, 2–5 may exit the protein pocket without reacting with the nucleophile. As a third option, the modified species may revert back to bound 1 via rapid intramolecular processes and, once more, not proceed in NEG.
Can Pi facilitate the formation of 2–5 from a glucopyranose? The first experimental observation consistent with Pi-facilitated formation of any (or all) of 2–5 from a glucopyranose in aqueous solution (Table ) is drawn from α-to-β glucopyranose inflection times. Both mono- and dianionic Pi facilitate the generation of electrophilic-modified species from a glucopyranose (dianionic being the greater facilitator). This observation is consistent with previous studies of glucose mutarotation by Bailey, Fishman, and Pentchev (Citation1970). Second, α- to β-G6P inflection times indicate that modification of the original structure to an electrophilic species takes place more rapidly than does Pi facilitated α-glucopyranose to β-glucopyranose conversion. Interestingly, G6P is known to glycate β-Val1 of HbA much more efficiently than does glucose (Haney & Bunn, Citation1976). Third, G1P decreased α- to β-glucopyranose inflection times by a factor of 10, likely because the phosphate on G1P is available for bimolecular reaction, playing a role similar to that of Pi.
In an effort to attain further mechanistic insight regarding Pi-facilitated α- to- β-glucopyranose interconversion, aqueous solutions at or near neutral pH with glucose:Pi molar ratios of 5:1 and 20:1, reflecting the relative intracellular concentrations of glucose in human erythrocytes (3–4 mmol/L, Santin et al., Citation2013) and Pi (0.5–0.8 mmol/L, Bevington et al., Citation1986) were prepared and inflection time experiments were performed. The inflection times for α- to- β-glucopyranose interconversion were much shorter than the benchmark (270 min), even when the HPO4−2 anion concentration is 1/20th that of the sugar. That is, a rapid, reversible glucopyranose/Pi interaction occurred whereby interconversion is facilitated within the lifetime of a transient interaction, and this was true even at physiologically relative concentrations.
Validation that Pi-facilitated formation of reactive electrophiles is the result of Pi structural effect and not a pH effect. Multiple reagents were evaluated for mechanistic purposes to discern whether inflection times are governed by effector reagent structure or whether there is a significant pH effect. From these studies, it is clearly a structural effect that is primarily manifest. First, large variation in inflection times were observed with different effector reagents at the same pH. Further, no trend exists in inflection time with pH (Table , Experiments 5–10). Second, at the common pH of 7.0, Bell (Citation1997) demonstrated significant reaction between glucose and amino acids in the presence of phosphate buffer but not in the presence of citrate buffer. Thus, at identical pH, different buffers promote the NEG process to different degrees—a structural effect rather than a pH effect. Third, the 31P chemical shift data are consistent with a reversible adduct between dibasic Pi and glucopyranose. Fourth, as computational validation (ab initio HF631G**) for the formation of the bridging adduct is 30 kcal/mol exothermic relative to isolated starting materials. Together, these data suggest that the primary structural effect of Pi is to bridge with a glucopyranose, enabling the generation of 2.
While these data support the contention that Pi does facilitate the formation of modified forms of glucopyranose, they do not directly enable the assignment of 2–5. Further, these results are relevant only if analogous reactions can take place between a Pi and a glucopyranose while concomitantly bound in a protein pocket.
Can Pi concomitantly bind with glucopyranose isomers in HbA? Computational modeling (Table ) predicts that exothermic, reversible binding of mono- and dianionic Pi with 1 occurs in known HbA glycation pockets in competition with reversible single-molecule binding of Pi and glucopyranose in those same pockets. X-ray crystallographic validation of concomitant binding of glucopyranose and Pi has been documented for human serum albumin (Wang et al., Citation2013).
Are there suitable geometries for the Pi-facilitated formation of 2–5 from a glucopyranose while concomitantly bound in the β-Val1 pocket of HbA? If so, what mechanistic possibilities exist? Computations predict an array of geometries of noncovalently bound Pi, glucopyranose, and proximate amino acid residues suitable for the formation of 2–5. Six mechanistic classes of reactions are geometrically predicted (Figures and ).
In mechanistic class I, Pi (mono- or dianionic) is geometrically able to bridge the bound glucopyranose (in which the Pi has two points of molecular interaction with the glucopyranose) by protonating the hemiacetal oxygen while deprotonating the anomeric OH in a concerted process, passing through 2 (Figure ). Support for Pi participation in a concerted acid/base reaction via a Pi-bridging interaction comes from HF-631G** ab initio computations (gas-phase). Each of the glucopyranose isomers (1) are predicted to bridge with either mono- or dianionic Pi. Moreover, this mechanism is analogous to that proposed for the mutarotation of glucose in aqueous solution at physiological pH in which water is the bridging agent (Silva, da Silva, & da Silva, Citation2006).
In mechanistic class II, Pi can act as either acid or base in conjunction with a proximate amino acid residue (Figure ), which also reacts with the glucopyranose (1) and again passes through 2. The species acting as the base can be a Lys R-NH2, dianionic Pi, or neutral His. The species serving as the acid can be a Lys R-, monoanionic Pi, or His-H+. Regardless of the specific acid or base, this mechanism passes through transient electrophile 2. Each variation in this mechanistic class is reasonable because analogous mechanisms (both stepwise, involving discrete intermediates and concerted, not involving discrete intermediates) are observed for the enzyme galactose mutarotase (Thoden et al., Citation2003). Irrespective of how 2 is generated or whether nucleophilic attack takes place as 2 is formed (concerted) or after (stepwise), a Schiff base can be generated. The Schiff base can then revert back to 1 or it can progress through NEG.
In mechanistic class III, the hemiacetal oxygen of a bound glucopyranose (1) is protonated by monoanionic Pi, His-H+, or Lys R- (Figure ). This process passes through transient electrophile 3, consistent with related chemistry proposed by Wang et al. (Citation2013) involving HSA. In mechanistic class IV, Pi is protonated by a Lys R-
or His-H+, which then protonates the hemiacetal oxygen of 1, again passing through 3 in a concerted fashion (Figure ). Johansen, Kiemer, and Brunak (Citation2006) have shown that a carboxylate is capable of abstracting the proton of an R-
amino acid residue in a concerted process in conjunction with nucleophilic attack, making it likely that Pi (mono- or dianionic) is also a sufficient base for this process.
In mechanistic class V, the geometries of various local minima enable the anomeric OH of a bound 1 to be protonated by monoanionic Pi, such that progression through transient electrophile 4 is possible (Figure ). Once more, either concerted (Numao et al., Citation2002) or stepwise processes (Qian, Citation2012) are theoretically possible. Mechanistic class VI also involves the protonation of the anomeric OH of bound 1 by monoanionic Pi, but here Pi undergoes concerted protonation by a Lys R- or His-H+ (Figure ). This process also passes through 4 and retains the original charge state of the Pi. Whether formed by mechanism V or VI, 4 can be attacked by a nucleophile to generate the cyclic glycosylamine that can continue through NEG. Another potential fate is for water to detach from 4 as a leaving group prior to nucleophilic attack in a stepwise process, passing through 5 (Figure ) and once more proceeding to a cyclic glycosylamine.
Can water facilitate the formation of bound 2–5 from a bound glucopyranose? A single water molecule can concomitantly bind in the β-Val1 pocket with either α- or β-glucopyranose and facilitate the formation of 2–5 via four mechanistic classes of reaction (fully described in Supplementary Figure 2) such that HbA1c can be generated. In each of these mechanisms, the β-Val1 R-NH2 serves a single role as the nucleophile. Concomitant binding of water and glucopyranose in either the Lys95 pocket or the Lys59/61/65/66 pocket possess geometries suitable for reaction involving Lys95 and Lys61, respectively. However, the amino acid residue to be glycated (Lys95 or Lys61) exists initially as an R- and thus must first act as an acid to protonate water, which must then protonate and ring open 1. The resulting R-NH2 amino acid can then theoretically react as a nucleophile. Such mechanisms, where a single amino acid residue plays two roles, are referred to here as dual-role mechanisms. Lys95 is not an experimentally observed glycation site (Delpierre, Vertommen, Communi, Rider, & Van Schaftingen, Citation2004), indicating that dual-role mechanisms are less viable than mechanisms that do not require a dual-role. As such, these types of mechanisms are predicted to be minor processes.
Can 2–5 be generated in the HbA β-Val1 pocket without the concomitant binding of an effector reagent? An effector reagent (ER) is any molecule that may facilitate or inhibit one or more mechanistic steps in NEG. Computations predict that a Lys R- can act as an acid, while, in a concerted fashion, a neutral histidine acts as a base to generate 2 (Supplementary Figures 3a and 4) which is attacked by the β-Val1 R-NH2 (in a concerted or stepwise fashion) as the first covalent step toward the ultimate generation of HbA1c. That histidine can abstract a hydroxyl proton in a concerted charge relay system is reasonable because far less basic species (e.g. carboxylate anions) are capable of this reaction in related systems (Stergiou, Foukis, Gkini, Bieth, & Papamichael, Citation2016; Thoden et al., Citation2003; Vladimirova et al., Citation2016; Wang et al., Citation2013; Zhang et al., Citation2001). Further, examples of His-H+ and β-Val1 or Lys R-
residues acting as acids in concerted mechanisms have been reported (Wang et al., Citation2013; Zhang et al., Citation2001).
Glycation in the absence of Pi (or water, or any other ER) is much less likely than glycation in the presence of Pi (or other ER). First, less than 10% of the computationally determined local minima meet the geometric requirement for this reaction. Therefore, the likelihood of HbA1c formation from 1 bound alone in the absence of an effector reagent is much lower than in the presence of an effector reagent (where 27% of minima can lead to glycation at Val1). This is significant in potentially explaining a pre-Amadori rate-determining Pi effect. Only one mechanism is predicted when glucopyranose binds alone in the β-Val1 pocket, whereas in the same pocket a glucopyranose in the presence of Pi enables six mechanistic categories comprising ca. 50 mechanisms. The likelihood of arriving at the geometry for the single mechanism for 1 alone is much lower than the probability of attaining suitable geometry for any of the ER-assisted mechanisms. Finally, the single mechanistic possibility can proceed only if the histidine is neutral (which is not always the case).
What is the impact of mechanistic diversity on site selectivity in the early, noncovalent stages of HbA glycation? The data addressing this question are summarized in Table S3.
Based upon the computational assessment of mechanistic possibilities in the β-Val1 pocket, we predict that Val1 will experience a high degree of glycation. This is in part due to its inherently high percentage as the R-NH2 form under physiological conditions (Matthew, Hanania, & Gurd, Citation1979), making it a readily available nucleophile. Further, Val1 is predicted to be in proximity to a relatively high concentration of bound 2–5 due to effective concomitant binding of both glucopyranose isomers with each species of Pi (especially dianionic Pi). In fact, Pi effects in the Val1 pocket are more pronounced than in any of the other HbA β-chain pockets. All of these factors contribute to the ability of β-Val1 to progress in NEG. While the glucopyranoses bind near Lys82, Lys82 is predominately in its R- form (Matthew et al., Citation1979) and hence, is non-nucleophilic and not able to react. That said, concomitant binding events of 1 with either species of Pi occur, and thus Pi can facilitate deprotonation to the nucleophilic amine. However, the Lys82 R-NH2 must then react as a nucleophile in a dual-role mechanism before 2–5 either revert back to 1 or dissociate from the pocket. This temporal demand may explain why little glycation at Lys82 is observed. No geometries predict a Lys8 glycation event. Lys132 and 144 [both predominately R-
ammonium ions (Matthew et al., Citation1979)] are also not predicted to undergo significant glycation because neither has the proper geometry to react during the binding of 1 and neither species of Pi binds to these residues in a manner that could lead to deprotonation and the generation of a nucleophile.
In the Lys59/61/65/66 pocket, glucopyranose alone binds near each lysine residue, although Lys59 and Lys66 are more accessible. However, like Lys82, these amino acid residues are predominately R- ammonium ions (Matthew et al., Citation1979) and are unlikely to proceed in NEG. Glucopyranose concomitant binding with Pi occurs near Lys59, Lys61, and Lys65 but not within reacting distance to Lys66. Overall, little glycation within this pocket is predicted.
Lys17 in the Lys17/120 pocket binds only α-glucopyranose with dianionic Pi. Lys120 binds neither glucopyranose isomer nor either Pi species independently, and no concomitant binding with dianionic Pi occurs with suitable geometry to react. Thus, little glycation of Lys120 or Lys17 is predicted.
In the Lys95 pocket, no glycation is computationally predicted because neither glucopyranose isomer nor either Pi species independently binds near Lys95 and no concomitant binding with dianionic Pi occurs with suitable geometry to react at this residue. The water-assisted Lys95 reaction is unfavorable as well, owing to it being a dual-role mechanism.
There is experimental support for each of our computational predictions regarding site selectivity for HbA glycation on the β-chain (Delpierre et al., Citation2004; Shapiro, McManus, Garrick, McDonald, & Bunn, Citation1979; Wang et al., Citation2013; Zhang et al., Citation2001). Overall, β-Val1 is the most glycated residue of HbA (reviewed in Delpierre et al., Citation2004), consistent with our computations. Lys82 (also in the β-Val1 pocket) however, is predicted to undergo limited glycation which is consistent with in vivo glycation for HbA (Delpierre et al., Citation2004; Zhang et al., Citation2001). This is especially interesting because when glyceraldehyde is used as a glycation surrogate, Lys82 glycates readily (Ito et al., Citation2011; Nacharaju & Acharya, Citation1992). Glyceraldehyde does not undergo an NEG process, but instead follows a nonenzymatic covalent protein modification (NECPM) mechanism and cannot be compared directly to NEG (Rodnick et al., Citation2017). Zhang et al. (Citation2001) reports that Lys8 combined with Lys17 is ca. 1% of total glycation. Our predictions are in agreement. Delpierre et al. (Citation2004) report Lys132 and 144 as the two lowest glycating residues, while Zhang reports as much as 6% glycation of Lys132. Zhang et al. (Citation2001) reports the combined glycation of Lys61/65/66 as 9% of all glycation events. Lys66 is reported for modest glycation by Shapiro and Delpierre, neither of whom indicates glycation for Lys65 (Delpierre et al., Citation2004; Shapiro et al., Citation1979). Our binding and geometric assessments predict that these residues would be glycated to a low degree. Lys95 is experimentally a non-glycating residue (Delpierre et al., Citation2004; Shapiro et al., Citation1979; Zhang et al., Citation2001), precisely what we predict. The consistency between computationally predicted site selectivity as determined by chemistry in the non-covalent stages and experimental observations suggests that diverse Pi effects in the non-covalent stages may impact the site selectivity of NEG for HbA (Table S3).
Which mechanism prevails in a given HbA pocket is based on multiple factors. One factor is the identity of the species bound in a given pocket (α- or β-glucopyranose, mono- or dianionic Pi, and/or water; concomitant or single species binding). Another is the specific geometries of the bound species relative to one another and to amino acid residues. The relative extent of mono- versus dianionic Pi is likely to impact the nature and extent of NEG because dianionic Pi facilitates many more mechanisms, while monoanionic Pi undergoes more exothermic concomitant binding with 1 and binds to more sites (Table ). Further, the identity and number of potentially acidic, basic, and/or nucleophilic amino acid residues are factors. Specifically, the presence of proximate N-terminal valine residues and lysine R-NH2 vs. R- affect mechanistic possibilities. The number, geometric availability, charge state, and resulting pKa of nearby histidines and carboxylates are also factors in determining which specific mechanisms are manifest. For a more complete assessment of these mechanisms, future studies should consider the persistence of reagents in various protein pockets, a concept referred to as residence time (Tummino & Copeland, Citation2008).
Site selectivity for NEG will likely be governed by the same variables that govern mechanisms. We propose the β-Val1 pocket of HbA is a clinically relevant “hot spot” because it can support multiple effector reagents and facilitates many more mechanistic possibilities than any other site. Further, β-Val1 is in an R-NH2 nucleophilic form within these variable mechanisms with a much higher probability than the competing lysine residues, which are most often non-nucleophilic R- ions (Matthew et al., Citation1979). Within the β-Val1 pocket, glycation of Val1 leading to HbA1c prevails over glycation of Lys82 and other nearby lysine residues in the same pocket based upon these same mechanistic arguments. The scope of this paper does not include the impact of the organic phosphate 2,3-bisphosphoglycerate (2,3-BPG), which is well known to bind to the same pocket (Arnone & Perutz, Citation1974). The effect of 2,3-BPG on the degree of mechanistic diversity, and its impact on site selectivity in the glycation of HbA is a focal point of our follow-up work.
Finally, we posit that HbA1c formation (and protein glycation in general) is slow because the predominant species bound (a glucopyranose, 1) binds in an unreactive form that must undergo reversible modification while bound. Further, the likelihood of the proper geometry for 1 to proceed in NEG in the absence of an ER is low. Facilitation by an ER enhances the overall rate, yet is a low probability event. First, concomitant binding events of a glucopyranose with an ER (i.e. Pi or water) are low probability events. Second, a suitable geometry to react is required for ER participation and these events are predicted to be rare (many more geometries are computationally observed that do not possess the proper geometry to react relative to those that do). Third, the resulting bound-modified glucose electrophile must react with a nucleophilic amino acid residue before either reversion to 1 or dissociation from the pocket occurs. Finally, a suitable nucleophile must be available with proper geometry within the lifetime of the electrophile. The requirement of meeting all four criteria simultaneously may contribute to why the glycation of HbA to HbA1c is a slow process.
4. Conclusions
In addition to Pi effects in the covalent stages of the NEG process, Pi effects are manifest in the early, noncovalent stages of NEG. Pi acts as an effector reagent by concomitantly binding with 1 and is predicted to react, while bound to HbA, to generate an array of transient electrophiles via multiple mechanisms. Six mechanistic classes and ca. 50 potential mechanisms, many of which may be concerted or stepwise, are predicted. Which mechanism prevails in a given HbA pocket is based (1) the identity of the species bound in a given pocket (α- or β-glucopyranose, mono- or dianionic Pi, and/or water; concomitant or single species binding); (2) the specific geometries of the bound species relative to one another and to amino acid residues; (3) the relative extent of mono- vs. dianionic Pi; and (4) the identity and number of potentially acidic, basic, and/or nucleophilic amino acid residues. The new Pi effects reported upon here in the non-covalent stages of the NEG process can help explain both HbA glycation site selectivity and the relatively slow rate of NEG.
4.1. Experimental procedures
4.1.1. NMR Experiments
All 1HNMR experiments were performed on a JEOL ECX-300 spectrometer. Reagents were all purchased commercially and used without further purification. Inflection time measurements were made by preparing a 1:1 M equivalent mixture of the α-glucopyranose to effector reagent at a composite concentration of 140 mmol/L in D2O. We also measured the time for α-G6P inflection to β-G6P in absence of any effector reagent except D2O. Reactions were followed as a function of time (ca. 5 min being the earliest time point based upon getting the NMR tube loaded, locked, and shimmed). From there, data were collected every 2–15 min, depending on the reaction progression. The doublet at 5.06 ppm for the proton on the anomeric carbon of the α-glucopyranose isomer diminishes with time as the doublet at 4.47 ppm for the proton on the anomeric carbon of the β-glucopyranose isomer correspondingly increases with time. The time that is required for the integration of the measured signals for the protons for the α-and β-glucopyranose isomer (at 5.06 and 4.47 ppm) to become equal is defined as the inflection time. As inflection times decrease, the rate of passage through the necessary transient electrophile (ring-opened isomer, Figure ) increases. An identical process was utilized to determine the inflection for α-G6P to β-G6P (following the doublet for the proton on the anomeric carbon of each species). These model reactions were used to assess the impact of an effector reagent to facilitate transient electrophile formation from a glucopyranose. This comparison between effector reagents was designed to give a qualitative understanding of relativistic effector reagent impact. The solution pH of each of the samples subjected to NMR analysis was determined with a pH meter. 31PNMR experiments were designed to follow chemical shift differences in mono- and/or dianionic Pi, as well as G6P, in their interactions with glucopyranoses. Reaction conditions and relative concentrations were identical to those used in our 1HNMR experiments. All 31PNMR were conducted at 20°C with chemical shifts measured against external standard phosphoric acid.
4.2.2. Computational substrates
Protein/substrate computations utilized Molecular Operating Environment (MOE, ver. 2014.9, Chemical Computing Group Inc., Montreal). The structures utilized in the computations for protein-substrate interaction were as follows: (a) the fully oxygenated HbA crystal tetramer 1GZX was obtained from the RCSB PDB (http://www.pdb.org/pdb/home/home.do), (b) the α- and β-glucopyranose isomers were obtained from Heterocompound Information Centre, Uppsala, (HIC-UP, http://xray.bmc.uu.se/hicup/) and (c) mono- and dianionic Pi and water were built with The PyMOL Molecular Graphics System (ver. 1.5.0.4 Schrödinger, LLC, http://www.pymol.org/) or made within MOE. Ab initio computations of substrate Pi adduct formation were performed in Spartan, version 1.0.3 (Wavefunction Inc. Irvine, CA USA).
4.2.3. Computational modeling to assess the binding of substrates to HbA
Within MOE, the HbA β-chain utilized was modified such that the Val1 was an R-NH2 amine. All lysines were R- ammonium ions. Single substrate experiments utilized the complete HbA protein. Structures that were exothermic beyond the limit of −2.5 kcal/mol (the binding exothermicity of water) were analyzed, with a cutoff distance of 5Å established for effective reaction within protein pocket(s) (Bobadilla, Nino, & Narasimhan, Citation2004; Ito et al., Citation2011). For concomitant binding studies, a glucopyranose was tether-docked in the pocket of interest and then a second substrate (i.e. Pi or water) was then inserted and energy minimized.
Supplemental data
Supplemental data for this article can be accessed at https://doi.org/10.1080/23312025.2018.1425196.
Funding
This work was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences [grant number P20GM103408]. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
| Abbreviations | ||
| 2,3-BPG | = | 2,3-bisphosphoglycerate |
| AGE | = | advanced glycation endproducts |
| G1P | = | glucose-1-phosphate |
| G6P | = | glucose-6-phosphate |
| HbA | = | human adult hemoglobin |
| HbA1c | = | human adult hemoglobin A1c |
| HSA | = | human serum albumin |
| MOE | = | Molecular Operating Environment |
| NEG | = | nonenzymatic glycation |
| NMR | = | nuclear magnetic resonance |
| Pi | = | inorganic phosphate |
OABI_1425196_Supplementary_Material.zip
Download Zip (858.4 KB)Additional information
Notes on contributors
Kenneth J. Rodnick
Our interdisciplinary research group consists of two principal investigators: a physiologist in the Biological Sciences Department (KJR) and an organic chemist in the Chemistry Department (RWH) at Idaho State University. Our research group also typically consists of 6–12 undergraduate students and several Master’s students from the Departments of Biological Sciences, Chemistry, Medical Laboratory Science, and Biomedical and Pharmaceutical Sciences. Our research focus is to better understand the bioorganic mechanisms associated with the nonenzymatic glycation of proteins. Our primary tools include computational methodology based upon crystal structures and model reactions followed by 31P and 1H nuclear magnetic resonance spectroscopy (NMR). Our goals are to predict what physiological reagents might be involved in the early stages of the nonenzymatic glycation of human hemoglobin (HbA) and to elucidate the mechanisms for the reactions that these physiological reagents have with HbA. This specific work highlights the importance of inorganic phosphate as a physiological reagent and its diverse role in accelerating the formation of glycated human hemoglobin to HbA1c.
Related Research Data
References
- Allen, K. N., Lavie, A., Glasfeld, A., Tanada, T. N., Gerrity, D. P., Carlson, S. C., … Ringe, D. (1994). Role of the divalent metal ion in sugar binding, ring opening, and isomerization by D-Xylose isomerase: Replacement of a catalytic metal by an amino acid. Biochemistry, 33(6), 1488–1494.10.1021/bi00172a027
- Arnone, A., & Perutz, M. F. (1974). Structure of inositol hexaphosphate–human deoxyhaemoglobin complex. Nature, 249(5452), 34–36.10.1038/249034a0
- Bailey, J. M., Fishman, P. H., & Pentchev, P. G. (1970). Anomalous mutarotation of glucose 6-phosphate. An example of intramolecular catalysis. Biochemistry, 9(5), 1189–1194.
- Bell, L. N. (1997). Maillard reaction as influenced by buffer type and concentration. Food Chemistry, 59, 143–147.10.1016/S0308-8146(96)00257-9
- Bevington, A., Mundy, K. I., Yates, A. J. P., Kanis, J. A., Russell, R. G. G., Taylor, D. J., … Radda, G. K. (1986). A study of intracellular orthophosphate concentration in human muscle and erythrocytes by 31P nuclear magnetic resonance spectroscopy and selective chemical assay. Clinical Science, 71, 729–735.10.1042/cs0710729
- Bobadilla, L., Nino, F., & Narasimhan, G. (2004, November 2). Predicting and characterizing metal-binding sites using support vector machines. In Proceedings of the International Conference on Bioinformatics and Applications (pp. 307–318). Fort Lauderdale, FL.
- Bookchin, R. M., & Gallop, P. M. (1968). Structure of hemoglobin A1c—Nature of the N-terminal beta chain blocking group. Biochemical and Biophysical Research Communications, 32, 86–93.10.1016/0006-291X(68)90430-0
- Brownlee, M.D, M. (1995). Advanced protein glycation on diabetes and aging. Annual Review of Medicine, 46, 223–234.10.1146/annurev.med.46.1.223
- Bryant, R. G. (1983). The NMR time scale. Journal of Chemical Education, 60, 933–935.10.1021/ed060p933
- Bunn, H. F., Haney, D. N., Gabbay, K. H., & Gallop, P. M. (1975). Further identification of the nature and linkage of the carbohydrate in hemoglobin A1c. Biochemical and Biophysical Research Communications, 67(1), 103–109.10.1016/0006-291X(75)90289-2
- Bunn, H. F., Haney, D., Kamin, S., Gabbay, K. H., & Gallop, P. M. (1976). The biosynthesis of human hemoglobin A1c. Slow glycosylation of hemoglobin in vivo. Journal of Clinical Investigation, 57(6), 1652.10.1172/JCI108436
- Bunn, H. F., & Higgins, P. J. (1981). Reaction of monosaccharides with proteins: Possible evolutionary significance. Science, 213(4504), 222–224.10.1126/science.12192669
- Clark, S. L. D., Santin, A. E., Bryant, P. A., Holman, R. W., & Rodnick, K. J. (2013). The initial noncovalent binding of glucose to human hemoglobin in nonenzymatic glycation. Glycobiology, 23, 1250–1259.10.1093/glycob/cwt061
- Delpierre, G., Vertommen, D., Communi, D., Rider, M. H., & Van Schaftingen, E. (2004). Identification of fructosamine residues deglycated by Fructosamine-3-kinase in human hemoglobin. Journal of Biological Chemistry, 279, 27613–27620.10.1074/jbc.M402091200
- Gil, H., Mata-Segreda, J. F., & Schowen, R. L. (1988). Proton transfer is not rate-limiting in buffer-induced non-enzymic glucation of hemoglobin. Journal of the American Chemical Society, 110, 8265–8266.10.1021/ja00232a065
- Gil, H., Peña, M., Vásquez, B., & Uzcategui, J. (2002). Catalysis by organic phosphates of the glycation of human hemoglobin. Journal of Physical Organic Chemistry, 15(12), 820–825.10.1002/poc.546
- Gil, H., Salcedo, D., & Romero, R. (2005). Effect of phosphate buffer on the kinetics of glycation of proteins. Journal of Physical Organic Chemistry, 18(2), 183–186.10.1002/(ISSN)1099-1395
- Gil, H., Vásquez, B., Peña, M., & Uzcategui, J. (2004). Effect of buffer carbonate and arsenate on the kinetics of glycation of human hemoglobin. Journal of Physical Organic Chemistry, 17, 537–540.10.1002/(ISSN)1099-1395
- Haney, D. N., & Bunn, H. F. (1976). Glycosylation of hemoglobin in vitro: Affinity labeling of hemoglobin by glucose-6-phosphate. Proceedings of the National Academy of Sciences, 73(10), 3534–3538.10.1073/pnas.73.10.3534
- Ito, S., Nakahari, T., & Yamamoto, D. (2011). The structural feature surrounding glycated lysine residues in human hemoglobin. Biomedical Research, 32(3), 217–223.10.2220/biomedres.32.217
- Johansen, M. B., Kiemer, L., & Brunak, S. (2006). Analysis and prediction of mammalian protein glycation. Glycobiology, 16(9), 844–853.10.1093/glycob/cwl009
- Kunika, K., Itakura, M., & Yamashita, K. (1989). Inorganic phosphate accelerates hemoglobin A1c synthesis. Life Sciences, 45, 623–630.10.1016/0024-3205(89)90048-9
- Lea, C. H., & Hannan, R. S. (1949). Studies of the reaction between proteins and reducing sugars in the “dry” state. Biochimica et Biophysica Acta, 3, 313–325.10.1016/0006-3002(49)90100-6
- Lee, C., Bagdasarian, M., Meng, M., & Zeikus, J. G. (1990). Catalytic mechanism of xylose (glucose) isomerase from Clostridium thermosulfurogenes. Characterization of the structural gene and function of active site histidine. Journal of Biological Chemistry, 265, 19082–19090.
- Lee, J. H., & Jeffrey, C. J. (2005). The crystal structure of rabbit phosphoglucose isomerase complexed with D-sorbitol-6-phosphate, an analog of the open chain form of D-glucose-6-phosphate. Protein Science, 14, 727–734.10.1110/ps.041070205
- Little, R. R., & Sacks, D. B. (2009). HbA1c: How do we measure it and what does it mean? Current Opinion in Endocrinology, Diabetes and Obesity, 16(2), 113–118.10.1097/MED.0b013e328327728d
- Matthew, J. B., Hanania, G. I., & Gurd, F. R. (1979). Electrostatic effects in hemoglobin: Hydrogen ion equilibria in human deoxy-and oxyhemoglobin A. Biochemistry, 18(10), 1919–1928.10.1021/bi00577a011
- Mohammad, A., Fraenkel-Conrat, H., & Olcott, H. S. (1949). The “browning” reaction of proteins with glucose. Arch. Biochem., 24, 157–178.
- Nacharaju, P., & Acharya, A. S. (1992). Amadori rearrangement potential of hemoglobin at its glycation sites is dependent on the three-dimensional structure of protein. Biochemistry, 31(50), 12673–12679.10.1021/bi00165a018
- Numao, S., Maurus, R., Sidhu, G., Wang, Y., Overall, C. M., Brayer, G. D., & Withers, S. G. (2002). Probing the role of the chloride ion in the mechanism of human pancreatic α-amylase. Biochemistry, 41(1), 215–225.10.1021/bi0115636
- Park, B., Holman, R. W., Slade, T., Murdock, M., Rodnick, K. J., & Swislocki, A. L. M. (2016). A biochemistry question-guided derivation of a potential mechanism for HbA1c formation in diabetes mellitus leading to a data-driven clinical diagnosis. Journal of Chemical Education, 93, 795–797.10.1021/acs.jchemed.5b00554
- Qian, X. (2012). Mechanisms and energetics for brønsted acid-catalyzed glucose condensation, dehydration and isomerization reactions. Topics in Catalysis, 55, 218–226.10.1007/s11244-012-9790-6
- Rodnick, K. J., Holman, R. W., Ropski, P. S., Huang, M., & Swislocki, A. L. (2017). A Perspective on Reagent diversity and non-covalent binding of reactive carbonyl species (RCS) and effector reagents in non-enzymatic glycation (NEG): Mechanistic considerations and implications for future research. Frontiers in Chemistry, 5, doi:10.3389/fchem.2017.00039
- Santin, A. E., Searle, A. J., Winston, V. D., Powell, M. S., Hardy, R. W., & Rodnick, K. J. (2013). Glycated hemoglobin is not an accurate indicator of glycemia in rainbow trout. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 165, 343–352.10.1016/j.cbpa.2013.04.012
- Shapiro, R., McManus, M., Garrick, L., McDonald, M. J., & Bunn, H. F. (1979). Nonenzymatic glycosylation of human hemoglobin at multiple sites. Metabolism, 28(4), 427–430.10.1016/0026-0495(79)90050-7
- Silva, A. M., da Silva, E. C., & da Silva, C. O. (2006). A theoretical study of glucose mutarotation in aqueous solution. Carbohydrate Research, 341, 1029–1040.
- Stergiou, P. Y., Foukis, A., Gkini, O. A., Bieth, J. G., & Papamichael, E. M. (2016). Kinetic and computational analysis of the reversible inhibition of porcine pancreatic elastase: A structural and mechanistic approach. Journal of Enzyme Inhibition and Medicinal Chemistry, 31(sup3), 131–139.10.1080/14756366.2016.1210137
- Stevens, V. J., Vlassara, H. E., Abati, A. L., & Cerami, A. N. (1977). Nonenzymatic glycosylation of hemoglobin. Journal of Biological Chemistry, 252(9), 2998–3002.
- Thoden, J. B., Kim, J., Raushel, F. M., & Holden, H. M. (2003). The catalytic mechanism of galactose mutarotase. Protein Science, 12(5), 1051–1059.10.1110/(ISSN)1469-896X
- Tummino, P. J., & Copeland, R. A. (2008). Residence time of receptor−Ligand complexes and its effect on biological function. Biochemistry, 47(20), 5481–5492.10.1021/bi8002023
- Vladimirova, A., Patskovsky, Y., Fedorov, A. A., Bonanno, J. B., Fedorov, E. V., Toro, R., … Raushel, F. M. (2016). Substrate Distortion and the Catalytic Reaction Mechanism of 5-Carboxyvanillate Decarboxylase. Journal of the American Chemical Society, 138(3), 826–836.10.1021/jacs.5b08251
- Wang, Y., Yu, H., Shi, X., Luo, Z., Lin, D., & Huang, M. (2013). Structural mechanism of ring-opening reaction by glucose by human serum albumin. Journal of Biological Chemistry, 288, 15980–15987.10.1074/jbc.M113.467027
- Watkins, N. G., Neglia-Fisher, C. I., Dyer, D. G., Thorpe, S. R., & Baynes, J. W. (1987). Effect of phosphate on the kinetics and specificity of glycation of protein. Journal of Biological Chemistry, 262(15), 7207–7212.
- Zhang, X., Medzihradszky, K. F., Cunningham, J., Lee, P. D., Rognerud, C. L., Ou, C. N., … Witkowska, H. E. (2001). Characterization of glycated hemoglobin in diabetic patients: Usefulness of electrospray mass spectrometry in monitoring the extent and distribution of glycation. Journal of Chromatography B: Biomedical Sciences and Applications, 759(1), 1–15.10.1016/S0378-4347(01)00196-7