
Abstract
Injecting anti-tumor necrosis factor (TNF)α antibodies into patient joints at the site of inflammation, inter-articular (IA) delivery, has demonstrated modest success for treatment of Spondyloarthritis (SpA), Rheumatoid Arthritis (RA), and osteoarthritis. However, IA delivery is not the treatment route of choice due to rapid clearance from the site of administration. Elastin-like polypeptides (ELPs) are reported to undergo phase transition; forming reversible aggregates above their transition temperature, which when injected into IA space have a 25-fold longer half-life compared to the soluble form. Here, we fused an ELP repeat to the C-terminus of each heavy chain of an anti-TNFα monoclonal antibody (mAb) and provide detailed characterization of the fusion IgG molecule. Expression and purification yielded homogenous protein confirmed by gels, hydrophobic-interaction chromatography, Capilary Electrophoresis (CE), Mass Spectrometry (MS), and analytical ultracentrifugation. The ELPs altered hydrophobicity and pI of the parent mAb and new elastic properties were imparted to the molecule; forming large stable complexes with a hydrodynamic radius of 40 nm above 39°C that dissociated into soluble, active monomer below 37°C. The fusion mAb retained its affinity and ability to neutralize TNFα as determined by surface plasmon resonance and cell-based assay, respectively, with equal potency to unmodified anti-TNFα mAb. Differential-scanning calorimetry studies show stabilization of adjacent CH2 and CH3 domains in the fusion molecule and the aggregated molecule was found to be fully functional after 7 days at 37°C. For the first time, we reveal architecture of an ELP-fusion mAb and binding to antigen using single-particle-transmission-electron microscopy. Unstructured ELP was visualized at the C-terminus and binding to antigen was shown at the N-terminus. Collectively, these studies indicate that it is possible to impart elastic properties to a monoclonal antibody while retaining purity, stability, and ability to effectively bind and neutralize antigen.
Public Interest Statement
Drawing inspiration from unique properties of molecules in nature, we engineered short elastin repeat motifs into a therapeutic protein. These short repeats make the fusion protein thermo-sensitive. Upon raising temperature, large stable aggregates are created and when the temperature is lowered, active monomer is released. Tumor necrosis factor (TNF), a cell cell-signaling cytokine, plays a central role in inflammation. This cytokine is produced by immune cells and upregulation results in diseases such as osteoarthritis (OA); a degenerative disease of the joints. Antibodies are proteins that can bind and block bad actors such as TNF and prevent them from harmful effects. However, injecting antibodies directly into joints to block TNF has shown modest success due to rapid clearance. Experimentation with bio-inspired materials, such as elastomeric proteins, provides therapeutic antibody-elastin fusion proteins with potentially longer lasting dwell time in the joint.
Competing interests
The authors declare no competing interests.
1. Introduction
The application of synthetic and natural biomaterials in biology and medicine dates back thousands of years with the use of gold, wood and glass in dentistry and prosthetics (Langer & Tirrell, Citation2004). Commercial use of synthetic polymers, such as polymethylmethacrylate in dentistry and cellulose acetate in dialysis tubing, can be traced to the early 20th century (Langer & Tirrell, Citation2004; Peppas & Langer, Citation1994). More recently, an emergence of new engineered materials is occurring that take their inspiration from nature. These bio-inspired materials are not only biocompatible and biodegradable, but can also possess unique properties, such as responsiveness to environmental cues (e.g. temperature and pH) (Langer & Tirrell, Citation2004; McMillan, Caran, Apkarian, & Conticello, Citation1999; Tatham & Shewry, Citation2000). One such class of biomaterial is elastomeric proteins from the building blocks of extracellular matrix (ECM) components. These elastomeric proteins are characterized by an ability to undergo significant change in form with added stress and to revert to their original state when the stress is removed (Tatham & Shewry, Citation2000). With recombinant DNA technology, it is possible to engineer environmentally responsive fusion proteins with repeat domains from elastomeric proteins (Elastin), which impart elastic behavior to the new molecule (MacEwan & Chilkoti, Citation2010).
Structural characterization of Elastin has revealed the presence of unique consensus tetrapeptides (VPGX), pentapeptides (VPGXG), and hexapeptides in the molecule, where X can be any amino acid other than proline and is termed the guest residue. These consensus sequences are responsible for the unique, thermoelastic behavior of Elastin (Nagapudi et al., Citation2002). Studies on these elastin-like polypeptides (ELPs) reveal thermo-sensitive behavior; soluble below the phase-transition temperature (Tt) while above the Tt, the ELPs adopt a more ordered structure (β-spiral) and begin to form large macromolecular complexes as a result of increased hydrophobic interactions (Kowalczyk, Hnatuszko-Konka, Gerszberg, & Kononowicz, Citation2014). The entire process is reversible, with solubility restored after decreasing the temperature below Tt. The phase temperature transition varies and can be tuned depending upon the identity of the guest residue, the ionic strength of the solution, as well as the overall mass of the polypeptide (Kowalczyk et al., Citation2014; McMillan et al., Citation1999). Increase in ionic strength and mass will decrease the Tt; hydrophobic guest residues lower Tt; and hydrophilic guest residues increase the Tt (Luginbuhl et al., Citation2017; McMillan et al., Citation1999). Sensitivity to pH can also be engineered into ELPs by substituting an ionizable amino acid into the guest position of the consensus peptides (McMillan et al., Citation1999; McMillan & Conticello, Citation2000).
ELPs can be recombinantly inserted or appended to therapeutic proteins at precise positions, imparting thermoresponsive properties while minimizing potential activity impairment. The novel use of ELPs in medicine has been investigated by a number of groups addressing unique challenges; such as, reduction of drug side effects by allowing fusion drug with polymer to accumulate at tumor sites or fusing cell-penetrating peptides to ELPs to improve therapeutic activity and applications to tissue engineering (MacEwan & Chilkoti, Citation2010). A single variable Camelidae anti-TNF heavy-chain domain (VHH), about 10 times smaller than conventional IgG, was fused to ELP to increase its size and extend half-life (Conrad et al., Citation2011). The molecule was expressed and purified from tobacco plant and shown to be active in vitro, and shown to block septic shock in mice. In a recent seminal study from the Chilkoti laboratory, the authors systematically explore the phase-transition behavior of GLP1-ELP (Luginbuhl et al., Citation2017). Sub-cutaneous deposition of GLP1-ELP-fusion protein resulted in slow release of active circulating drug for 10 days in mice and 17 days in monkeys. The remarkable potential of such a formulation was demonstrated by single treatment and glycemic control for 10 days in mouse models of diabetes. Earlier studies suggest that ELPs are non-immunogenic and non-pyrogenic (Urry, Parker, Reid, & Gowda, Citation1991); however, studies from the Chilkoti lab reveal non-neutralizing anti-drug antibodies (ADA) were found in their monkey studies (Luginbuhl et al., Citation2017).
Osteoarthritis (OA), a degenerative disease that affects joints, is a debilitating condition in a large section of the population, requiring long-term therapeutic intervention (Evans, Kraus, & Setton, Citation2014). Treatment of OA is primarily via chronic and oral administration of non-steroidal, anti-inflammatory drugs, which can result in GI bleeding and ulcers. Inter-articular (IA) delivery of corticosteroids and hyaluronan based products into diarthrodial joints offers some relief but has no disease-modifying effects (Evans et al., Citation2014; Hollander, Brown, Jessar, & Brown, Citation1951; Marshall, Citation2000). Localized delivery of small molecules and unmodified protein therapeutics is ineffective due to relatively rapid clearance from the synovial fluid. Small molecules and larger protein therapeutics are rapidly cleared from the joints via capillaries and the lymphatic system, respectively (Simkin, Citation1995; Simkin, Bassett, & Koh, Citation1995). The use of ELP-fusion proteins (ELPylation) for local delivery into diarthrodial joints was first pioneered by fusing interleukin-1 receptor antagonist (IL-1Ra) or soluble tumor necrosis factor antagonist (sTNFRII) to ELPs, which promotes formation of aggregating particles at physiologic temperature (Shamji et al., Citation2007, Citation2008). In both these studies, submicron-sized protein particles were reported after phase transition, creating local “depots” of the therapeutics. These depots are intended to increase dwell time in joint space and have been reported to increase dwell time 25 fold compared to the unmodified protein (Betre et al., Citation2006).
Tumor necrosis factor-alpha (TNFα) is a pro-inflammatory type II cytokine with diverse biological roles. TNFα is produced by both immune cells (T-cells, B-cells, monocytes/macrophages, and dendritic cells) and non-immune cells (osteoclasts, endothelial cells, adipocytes, and fibroblasts), and is a clinically validated target for the treatment of rheumatoid arthritis (RA), psoriatic arthritis, spondyloarthritis (SpA), Crohn’s disease, ulcerative colitis, chronic psoriasis, hidradenitis suppurativa, and juvenile idiopathic arthritis (Blanco et al., Citation2009; Brehm, Daniels, & Welsh, Citation2005; Cawthorn & Sethi, Citation2008; de Waal Malefyt, Abrams, Bennett, Figdor, & de Vries, Citation1991; Havell & Rogerson, Citation1993; Ho et al., Citation2001; Imaizumi et al., Citation2000; Kriegler, Perez, DeFay, Albert, & Lu, Citation1988; Nakao, Fukushima, Kajiya, Ozeki, & Okabe, Citation2007; Perez et al., Citation1990; van der Zee, Laman, de Ruiter, Dik, & Prens, Citation2012; Wajant, Pfizenmaier, & Scheurich, Citation2003; Williamson, Carswell, Rubin, Prendergast, & Old, Citation1983). A number of approved anti-TNFα therapeutics such as Adalimumab, Infliximab, and Golimumab (monoclonal antibodies), Etanercept (TNF-R2-Fc fusion) and Certolizumab (Fab fragment linked to polyethylene glycol) bind TNFα and block its interactions with receptors. Clinical studies on injecting anti-TNFα agents such as etanercept and infliximab into patient joints have reported successes for treatment of SpA, RA, and OA (Fiocco et al., Citation2010; Fioravanti, Fabbroni, Cerase, & Galeazzi, Citation2009; Fisher & Keat, Citation2006; Sakellariou, Kakavouli, & Chatzigiannis, Citation2006; van der Zee et al., Citation2012). However, IA delivery of these anti-TNFα agents has not resulted in treatment of choice primarily due to short residence time, or dwell time, in the joint space (Evans et al., Citation2014).
Here, we engineer elastin repeat sequences on the C-terminus of the heavy chain of an anti-TNFα monoclonal antibody and apply rigorous analytical methods to evaluate purity, physicochemical characteristics, activity, structure, and stability of the fusion molecule. The purified protein was shown to be homogeneous upon applying many orthogonal methods. The ELPs contributed to an increase in hydrophobicity and pI, and imparted elastic properties to the fusion molecule; reversible aggregates form above the transition temperature (Tt) of 39°C and active, monomeric protein restored below the Tt was shown to bind and neutralize soluble TNFα. Transmission electron microscopy images confirmed the Y-shaped structure of an antibody as well as binding to TNFα. Two other studies were conducted to evaluate systemic exposure. First, we engineered a mutation (H435C) in the Fc region to disrupt binding to FcRn (Baker et al., Citation2011; Rath, Baker, Pyzik, & Blumberg, Citation2014; Rath et al., Citation2013; Shields et al., Citation2001). The anti-TNFα ELP with the Fc mutation showed a significantly reduced rat half-life of less than 1 day. We also evaluated uptake of the fusion mAb into professional antigen-presenting cells (dendritic cells) and found no difference in uptake when compared to the parental mAb.
The results presented in this report with an anti-TNFα ELP mAb are a foundation for building on a new generation of fusion ELP mAbs, or other formats, that are stable, active, responsive to cues in local environment, and, with the FcRn mutation, cleared rapidly from circulation. More detailed studies are warranted to identify the appropriate ELP sequences for IA delivery, calculate residence time in the IA space, and demonstrate pharmacodynamics effect of the ELP-fusion protein.
2. Results
2.1. Design of fusion protein (anti-TNFα-ELP fusion mAb)
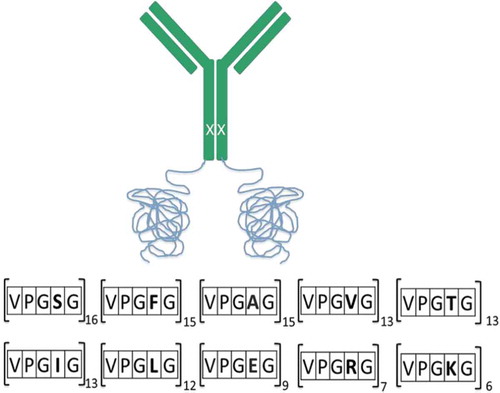
In Figure , we have an illustration of the fusion protein that was engineered to have Elastin repeats (ELPs) and a point mutation on the heavy chain (H435C) to abrogate binding to the neonatal Fc receptor. The anti-TNFα monoclonal antibody was cloned into a pHYBE expression vector with insertion of ELP gene sequences at the C-terminus; encoding for VPGXG119, adding approximately 50 kDa to each heavy chain. Guest residues (X) which were chosen for this study had a large number of hydrophobic (60% of valine, alanine, phenylalanine, isoleucine, and leucine) and fewer polar (20% of serine and threonine) and charged (20% of lysine, arginine, and aspartic acid) residues. Both the polar and charged residues were randomly interspersed among the hydrophobic residues to mitigate patches of hydrophobicity and charge on the fusion protein. A kappa light chain was used and the fusion molecule was expressed by transient transfection in HEK293-EBNA cells.
Figure 1. An illustration of the fusion mAb with insertion of ELP at the C-terminus (VPGXG119) of the heavy chain. Polar (20%) and charged (20%) residues were randomly interspersed among the hydrophobic (60%) residues to mitigate patches of hydrophobicity and charge on the fusion protein. A single-point mutation (H435C) was engineered in the heavy chain to disrupt binding to the neonatal receptor (depicted as X on HC).
2.2. Purification and physicochemical characterization of anti-TNFα-ELP mAb
Conventional Protein A chromatography, as the first capture step, was not possible as the H435C mutation, which knocked out FcRn binding, also impacted binding to the Protein A resin. We modified our purification scheme and the fusion mAb was captured from harvest material using cation-exchange chromatography then followed by anion-exchange chromatography and finally size-exclusion chromatography (SEC). Figure is an Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) gel with lanes 1–3 showing relative purity of the non-reduced fusion protein after each chromatography step. We observe enriched protein after the SEC run. Under reducing conditions (lanes 5 and 6), we confirmed purity and size of the fusion protein relative to parental monoclonal antibody. The expected size of the heavy chain of the fusion protein was shown to be about 100 kDa. We also used mass spectrometry, after tryptic digestion, to confirm the complete sequence of the fusion protein (Supplemental Figure ).
Figure 2. (a) SDS-PAGE gel with lanes 1–3 showing purity of the non-reduced fusion protein after each chromatography step. Lanes 5 and 6 show the fusion mAb and parental mAb, respectively, run under reducing conditions. (b) Size-exclusion chromatography (SEC) analysis of the fusion protein. The fusion protein was also incubated at 1:1 molar ratio with human TNFα and rerun on the SEC column. (c) Evaluation of the antibody and fusion protein hydrophobicity by hydrophobic-interaction chromatography (HIC) on a TOSOH Butyl-NPR column. (d) Evaluation of isoelectric point (pI) by imaged capillary isoelectric focusing (icIEF). (e) Use of sedimentation velocity, a solution based analytical methodology, to confirm the correct size of the fusion protein.
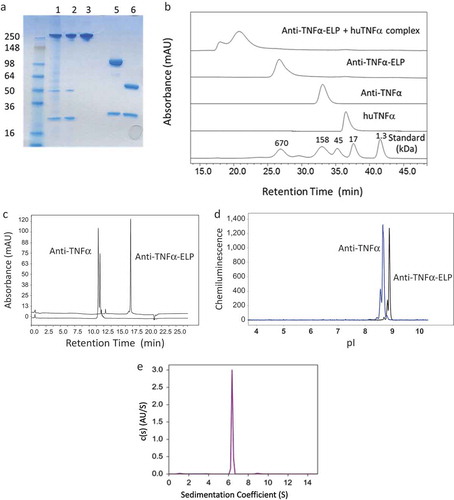
Size heterogeneity of the fusion protein and its ability to bind antigen in native solution conditions was evaluated by SEC. Figure shows SEC chromatograms for both mAb and ELP-fusion mAb. Though the antibody has an elution profile consistent with its expected molecular weight (~150 kDa), the fusion protein showed an elution position of a much larger protein (700 kDa) than the anticipated 250 kDa. We believe this is due to the random-coil nature of the engineered-ELP repeat as well as its high degree of hydrophilicity and interaction with water, which increased its apparent hydrodynamic size. In another study aimed at verifying that the ELP-fusion protein was homogeneous and of correct molecular weight, we used sedimentation velocity (AUC) and confirmed a total mass of 240 kDa (Figure ).
To evaluate the ability of the fusion protein to bind antigen, we incubated with human TNFα at 1:1 molar ratio and reran on the SEC column. The SEC profile in Figure showed large protein complexes eluting in the void volume that appeared to be fusion protein bound to antigen; no free fusion protein or human TNFα was observed.
We expected that insertion of hydrophobic and charged residues as guest residues in the pentapeptide repeat (VPGXG) would alter both hydrophobicity and isoelectric point (pI) or charge characteristics of the fusion mAb. To evaluate relative change in hydrophobicity, we used hydrophobic-interaction chromatography (HIC) and in Figure , we show that the parent anti-TNFα mAb has a retention time of approximately 11 min; whereas, the retention time of the fusion protein was increased to 17 min. The heterogeneity observed with the parental anti-TNFα mAb is C-terminal lysine processing that is not found in the fusion molecule. Using capillary isoelectric focusing (cIEF) we showed that the pI of the unmodified protein was 8.66, whereas the fusion molecule was increased to 8.89 (Figure ).
In summary, we reveal correct size, homogeneity, purity, and physicochemical properties (charge and hydrophobicity) of the ELP-fusion protein using orthogonal techniques such as SDS-PAGE, AUC, HIC, and cIEF analysis. Further, using mass spectrometry, we confirmed the correct sequence of the molecule with no evidence of splice variants.
2.3. Characterization of the phase-transition behavior of anti-TNFα-ELP mAb
Dynamic light scattering (DLS), a technique for determining size distribution, was used to evaluate the phase-transition behavior of the fusion protein. The phase-transition temperature is defined as the temperature with a 50% increase of the maximal change recorded in the hydrodynamic radius (Rh) of the molecule.
In Figure , we show Rh data for the anti-TNFα-ELP mAb at 0.5 mg/mL in 1×PBS. The temperature was steadily raised in increments of 0.065°C per minute from room temperature (25°C) up to 50°C, followed by a steady decrease back to 25°C at the same rate; all in one temperature-looping experiment. During the temperature ramp-up, the anti-TNFα-ELP mAb transitioned to a higher-order structure with an Rh value of approximately 34 nm at 50°C. The high-order protein ensemble displayed a homogeneous size distribution and a low dispersity index. After the temperature reached 50°C, the cooling step followed and temperature was then decreased back to 25°C. During the cooling time, a hysteresis like transition was observed as indicated in Figure . The upward Tt for formation of the large complex was estimated to be about 39–40°C and the reverse Tt for reverting back to soluble protein was determined to be about 37°C.
Figure 3. (a) DLS data were collected on a DynaPro Plate Reader. The anti-TNFα-ELP mAb concentration was at 0.5 mg/mL and temperature was steadily increased at 0.065°C/min. The temperature was also steadily decreased back to its start temperature of 25°C. (b) An illustration of possible transition states of the fusion protein.
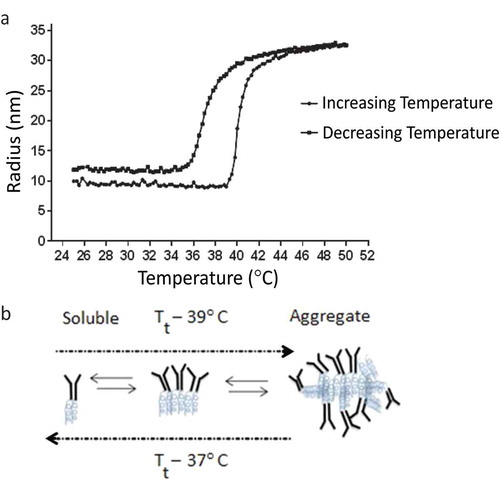
In a second study, we lowered the concentration of the anti-TNFα-ELP mAb protein to 0.1 mg/mL to control for solution viscosity and better estimate particle size using the Stokes-Einstein equation (Shamji et al., Citation2008). Data were collected on a Wyatt, DynaPro Nanostar instrument at intervals of 2°C and samples were allowed to equilibrate and reach the new temperature before the next reading was recorded. In this study, we observed an upward Tt of about 41°C and the aggregated protein complex plateaued at an Rh value of 40 nm at 55°C. Upon reversing the temperature, the aggregated complex reverted to its original radius of 10 nm and the reverse Tt was observed to be 37°C (data not shown). Figure is a cartoon that speculates how the fusion protein assembles and forms higher-order structures that can revert, upon lowering the temperature, to a fully functional fusion protein that retains the ability to bind TNFα.
2.4. Evaluation of thermal stability, binding, and neutralization properties of the anti-TNFα ELP fusion mAb
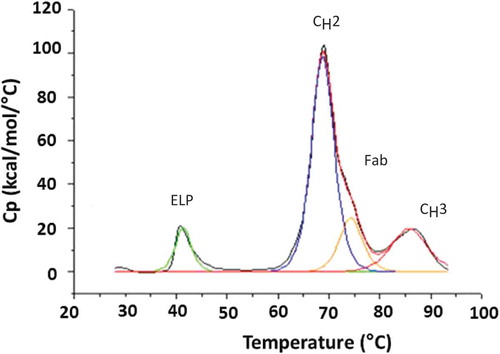
Thermal stability is a measure of the temperature-induced domain unfolding that is monitored by differential scanning calorimetry (DSC). The fusion protein was observed to have an early thermal unfolding temperature (Tm) of 41°C (Figure ) that is likely contributed from the ELP domain. The assignments of CH2, CH3, and Fab domains for the parent mAb and fusion molecule are based on literature and were not confirmed using independently purified domains (Ionescu, Vlasak, Price, & Kirchmeier, Citation2008). Both the unmodified mAb and fusion protein show a comparable onset of unfolding temperature (Tonset) of 60°C for the putative CH2 domain. The Tm and apparent enthalpies (app ΔH) for each domain are shown in Table . The higher apparent enthalpy observed with the parent mAb is typically associated with the Fab domain, given its large size and the higher energy, which is required to unfold that domain. With the addition of the ELP domain to the C-terminus, we observe a higher apparent enthalpy (see Table ) associated with the assigned CH2 and CH3 domains in the fusion molecule.
Table 1. Differential scanning calorimetry studies on parent anti-TNFα and anti-TNFα-ELP fusion mAb
Figure 4. Representative DSC scan for the anti-TNFα-ELP fusion mAb. Analysis was performed on a MicroCal VP-DSC instrument. Samples were diluted to 0.5–1 mg/mL in PBS buffer pH 7.4 and subjected to a temperature ramp from 25°C to 90°C at a rate of 1°C/min. The assignments of CH2, CH3, and Fab domains are based on literature and not confirmed using independently purified domains.
After purification of the anti-TNFα-ELP mAb, we evaluated it’s binding to TNFα and calculated the equilibrium dissociation constant (KD) before formation of supra-molecular complexes and after the complex was formed and reverted to form the soluble species. The sensorgrams are shown in Figure , with results summarized in Table , and the observed KD was comparable to unmodified mAb indicating that the ELP domain on the Fc portion of the molecule did not interfere with the Fab portion of the molecule and binding to antigen. The association and dissociation constant for the fusion protein was comparable to the parent mAb.
Table 2. Surface plasmon resonance for the binding of anti-TNFα and anti-TNFα-ELP fusion mAb to human TNFα
Figure 5. Sensorgram for binding of anti-TNFα antibodies to recombinant human TNFα obtained on a Biacore T200 instrument. (a) and (d) *Stable complex were formed at 40°C before measuring affinity at RT. (c) and (f) **Stable complex formed at 40°C and then kept for 7 days at RT before measuring affinity. Goat anti-human IgG Fc antibodies were immobilized via amino coupling on the chip surface. Antibodies, diluted to a concentration of ~ 0.5 − 1.0 µg/mL in HBS-EP+ buffer, were captured on flow cell and recombinant human TNFα injected for 5 min at a flow rate of 50 µL/min with continuous flow of the running buffer at a flow rate of 50 µL/min. Biacore T200 Evaluation Software version 2.0 was used to fit the data from the TNF binding studies to a 1:1 kinetic model.
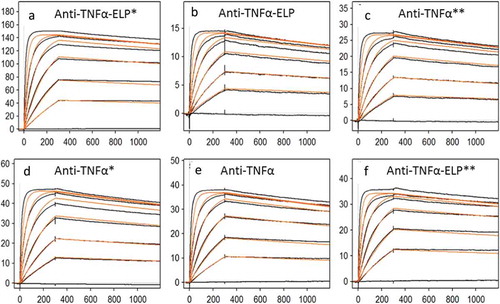
Stability of the fusion protein was evaluated at three different conditions; evaluation of affinity to TNFα at room temperature (RT), raising the temperature to 40°C to form stable complex, and then measuring affinity to TNFα at RT and finally by raising the temperature to 40°C to form stable complex and then storing the molecule at RT for 7 days before evaluating its affinity to TNFα. The sensorgrams are shown in Figure and the calculated KD summarized in Table . Under all three conditions evaluated, the fusion molecule retained binding properties to TNFα.
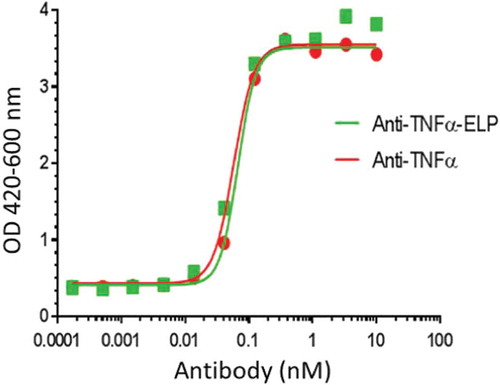
The addition of a simple and rapid bioassay with high sensitivity and good reproducibility is important for the analysis of neutralization of TNFα cytotoxicity. Thus, bioactivity of the anti-TNFα-ELP mAb was also assayed against TNFα mediated L929 cell cytotoxicity (Figure ) and the results are summarized in Table . The IC50 for neutralization of TNFα obtained for the unmodified mAb using either human or cynomolgus monkey TNFα was comparable to anti-TNFα-ELP mAb.
Table 3. In-vitro bioactivity data (IC50) of parent anti-TNFα or anti-TNFα-ELP fusion mAb assessed against human or cyno TNFα-mediated L929 cytotoxicity
Figure 6. Neutralizing assay on TNFα induced cytotoxicity in mouse L929 cells: Anti-TNFα-ELP fusion mAb and anti-TNFα mAb were serially diluted and pre-incubated with recombinant human TNFα (2.9 × 10−12 M final concentration). The pre-incubated samples were added to the murine fibrosarcoma cell line L929. Viability was quantified using WST-1 reagent. The plates were read at OD 420–600 nm on a Spectramax 190 ELISA plate reader (Molecular Devices, Sunnyvale, CA). The IC50 value was determined by plotting the OD420–600 values vs. the concentration of mAb-ELP using a non-linear four parameter curve fit using Graphpad Prism 5.
2.5. Structural analysis of anti-TNFα-ELP mAb alone and bound to antigen
Although it is challenging to discern detailed structure of a molecule from unprocessed transmission EM images, the signal-to-noise ratio can be greatly improved by identifying, aligning, classifying, and averaging particles in similar conformations (Correia et al., Citation2013). The resultant two-dimensional (2D) class averages of the anti-TNFα-ELP fusion mAb show “Y”-shaped particles consistent with those of antibodies (Figure ). The anti-TNFα-ELP fusion mAb has two arms (Fabs) and most averages show a typical ~7 nm Fc. However, some averages show a longer ~10–13 nm Fc with a clear additional area of density, about 4 nm in size proximal to the Fc (red arrow in Figure ) that most likely corresponds to the elastin attached to the Fc. The additional area of density can appear immediately adjacent to the Fc or at a slight distance and connected by weak density (orange arrow in Figure ). The overall density of the Fc region and ELP domain in the 2D class averages was weaker and less defined compared to the Fab arms, suggesting high flexibility in this region.
Figure 7. (a) Exemplary class averages (>200) denoted with possible interpretations of structure and orientation. Many class averages show a typical Fc size of about 7 nm. However some averages show a longer ~10–13 nm Fc with a clear additional area of density after the Fc (red arrow) that most likely corresponds to the elastin attached to the Fc. The additional area of density can appear immediately adjacent to the Fc or at a slight distance and connected by weak density (orange arrow). (b) Exemplary class averages of human TNFα show a typical size of about 4.9 nm. (c) Exemplary class averages of anti-TNFα-ELP fusion mAb incubated with huTNF-α showed particles that resembled dimers with two antibodies bound to two TNFα trimers; facing each other Fab to Fab in a diamond formation and some averages that resembled an antibody monomer bound to TNFα. The Fc portion of the antibodies was not clearly visible and resembled a faint blur; most likely due to the high flexibility of the Fc.
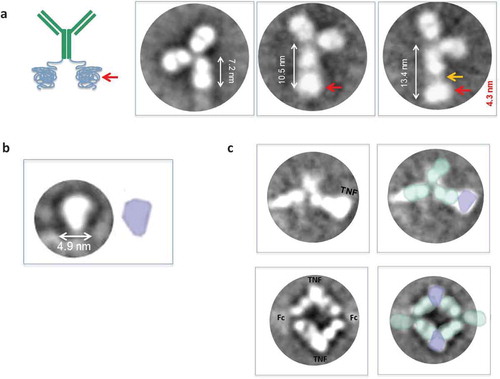
The 2D class averages of the TNFα trimer revealed particles of about 4.9 nm in size; individual subunits could not be distinguished (Figure ). Incubation of anti-TNFα-ELP mAb with TNFα (1:4) showed few particles that resembled antibody monomers or free TNFα in the sample. However, 2D averages showed particles that resembled dimers and some particles that resembled an antibody monomer bound to TNFα (Figure ). Averages that resembled an antibody dimer appeared to show two antibodies bound to two TNFα trimers facing each other Fab to Fab in a diamond formation. The Fc portion of the antibodies was not clearly visible and all attempts at imaging resembled a faint blur; very likely contributed from extreme flexibility.
3. Pharmacokinetic studies
Large molecules are cleared from IA space via the lymphatic system and enter circulation. The presence of large immune complexes of the anti-TNFα-ELP fusion mAb bound to TNFα in circulation is of some concern. To reduce systemic exposure, we also engineered a mutation in the Fc region (Histidine-435) of both parent mAb and ELP fusion mAb that disrupted binding to FcRn at pH 6. Neonatal receptor FcRn, a membrane associated receptor, has an important role in recycling and increasing the half-life of both IgG and serum albumin proteins; with each molecule having distinct binding sites on FcRn. Mutations designed to disrupt FcRn binding to the Fc region of an IgG molecule, such as H435C, will reduce recycling and half-life of IgG molecules (Shields et al., Citation2001). The neonatal Fc receptor is also reported to play a role in the presentation of peptides in the MHC complex by antigen-presenting cells. The FcRn mutation may reduce immunogenicity.
To evaluate the impact of the FcRn mutation on PK, we carried out studies in Sprague Dawley rats after intravenous (IV) administration. IV dosing (5 mg/kg) of the anti-TNFα mAb (H435C) revealed biphasic decline and T1/2 of less than 5 days (Supplemental Figure and Table ). The ELP fusion mAb, with FcRn mutation was dosed equimolarly (8.33 mg/kg) to the anti-TNFα mAb and exhibited a more rapid monophasic decline and a T1/2 of less than 1 day. Further studies are warranted to understand this rapid decline. However, these data provide evidence that the ELP-fusion protein entering the systemic circulation can be rapidly cleared.
4. Discussion
Elastic properties were successfully engineered into an anti-TNFα monoclonal antibody with the fusion protein demonstrating reversible phase transition. Above a Tt of 39°C, the molecule formed stable large complexes that plateaued at a Rh of about 40 nm. Upon lowering the temperature, soluble active monomer was formed at a lower Tt of 37°C. With the fusion protein, we retained full binding and neutralization potential to TNFα without compromising the overall stability of the molecule. This achievement is different from studies carried out with sTNFRII fused to ELP where the authors observed considerable destabilization of their fusion protein with loss in binding affinity and potency (Shamji et al., Citation2008). Thermal stability of our fusion molecule was comparable to the parent mAb; the ELP domain appeared to increase the apparent enthalpy of both the CH2 and CH3 domain of the fusion molecule.
In our studies, we were encouraged by good expression levels of the fusion protein when the ELPs were engineered on the heavy chain. Attempts to engineer ELPs on the light chain showed poor expression (results not shown). Purification of the fusion protein included cation- and anion-exchange chromatography followed by SEC, which provided high-quality material with low endotoxin levels. To mitigate patches of hydrophobic and charged residues on the molecule, we randomly inserted a guest residue in VPGXG in the ELP at a 60:20:20 ratio of hydrophobic, polar, or charged amino acids. The overall hydrophobicity and pI of the molecule was not substantially different from the unmodified mAb. Analytical evaluation of the anti-TNFα-ELP by mass spectrometry confirmed primary sequence coverage and other technologies such as analytical ultracentrifugation and differential scanning calorimetry were supportive of the correct secondary and tertiary structure. The ability to bind and neutralize TNFα further confirmed structural integrity of the molecule.
Studies have demonstrated that ELP-fusion proteins injected into IA space prolong dwell times with a half-life of 3.5 days (Betre et al., Citation2006). Literature report suggests that ELPs have potential to be non-immunogenic and have decreased serum exposure and accumulation in peripheral organs (Urry et al., Citation1991). However, studies from the Chilkoti laboratory reveal non-neutralizing ADA response to GLP1-ELP in their monkey studies. Our studies using dendritic cells show that the fusion protein is internalized; albeit with a lower efficiency than the unmodified mAb (supplemental Figure ). To facilitate systemic clearance of the fusion protein and reduce the possibility of an immunogenic response, we engineered a mutation in the Fc region (Histidine-435) on the IgG molecule that disrupts binding to FcRn (Shields et al., Citation2001). Pharmacokinetic (PK) evaluation of the anti-TNFα molecule with the FcRn mutation revealed a short half-life of 110 h. Interestingly, the half-life of the fusion protein was reduced even further to 20 h, and further studies are warranted to understand the enhanced clearance. The question that still remains unanswered is whether prolonged dwell time of anti-TNFα-ELP fusion mAb in joint space can effectively treat inflammation. Future studies will select the appropriate ELP and compare pharmacodynamics (PD) effects of the conjugated molecule to the unmodified therapeutic after delivery into IA space
5. Materials and methods
5.1. Expression of recombinant anti-TNFα-ELP fusion mAb
Anti-TNFα-ELP heavy chain and light chain were created by de-novo gene synthesis (Blue Heron, Bothell, WA). The following amino acid sequence of ELPs was added to the C-terminus of the heavy chain:
VPGSGVPGVGVPGTGVPGFGVPGAGVPGIGVPGEGVPGVGVPGRGVPGFGVPGTGVPGIGVPGAGVPGFGVPGSGVPGIGVPGLGVPGTGVPGVGVPGEGVPGFGVPGKGVPGLGVPGSGVPGIGVPGAGVPGFGVPGTGVPGSGVPGRGVPGLGVPGSGVPGFGVPGTGVPGVGVPGKGVPGFGVPGAGVPGIGVPGSGVPGLGVPGTGVPGVGVPGEGVPGFGVPGRGVPGLGVPGIGVPGTGVPGAGVPGSGVPGVGVPGRGVPGFGVPGTGVPGTGVPGAGVPGIGVPGVGVPGTGVPGAGVPGFGVPGRGVPGSGVPGVGVPGSGVPGLGVPGKGVPGIGVPGEGVPGVGVPGAGVPGSGVPGIGVPGLGVPGRGVPGVGVPGAGVPGEGVPGLGVPGSGVPGAGVPGKGVPGFGVPGAGVPGIGVPGEGVPGFGVPGAGVPGLGVPGVGVPGKGVPGSGVPGIGVPGLGVPGTGVPGFGVPGTGVPGKGVPGIGVPGAGVPGVGVPGSGVPGEGVPGIGVPGAGVPGLGVPGEGVPGSGVPGRGVPGFGVPGLGVPGSGVPGTGVPGFGVPGEGVPGVGVPGSGVPGAG
The expression constructs were cloned into pHYBE vector and expressed by transient transfection in HEK293-EBNA cells. The cells were cultured in a 10L Wave bag at 0.65–0.75 × 105 cells/mL in Freestyle 293 expression medium (Invitrogen, Carlsbad, CA) and grown overnight at 8% CO2 and 37°C. Next day, the cell density was adjusted to 0.8–1.2 × 106 cells/mL before transfection. For transfection, 5 mg of plasmid DNA (3 mg light chain and 2 mg heavy chain) was diluted into 350 mL of Optimem (Thermo Scientific, Waltham, MA) in a flask and 20 mL of polyethyleneimmine (PEI) (Polysciences, Warmington, PA) at a concentration of 0.5 mg/mL. After 5-min incubation at room temperature, DNA and PEI were mixed together and incubated at room temperature for 15 min. DNA:PEI complex was then added into the 10 L culture and incubated at 37°C, 8% CO2 with a rocking angle of 19°. Cells were fed at day 1 with 5 g/L tryptone N1 (Organotechnie, La Courneuve, France) in Freestyle 293 medium. After 7 days, cells were harvested and passed through a 0.45 micron cellulose acetate filter (Corning, NY) and the protein was then purified from the supernatant.
6. Purification of fusion protein
Cell supernatant was diluted 1:3 with distilled water and loaded on a SP Sepharose HP column (GE Healthcare, Chicago, IL) equilibrated in 50-mM phosphate containing 50-mM NaCl pH 7.2. After loading, the column was washed with phosphate buffer containing 50-mM NaCl. Protein was eluted with 50-mM phosphate containing 150-mM NaCl. The elution was diluted 1:3 with 50-mM phosphate pH 7.2 and the unbound fraction collected from a HiTrap Q FF (GE Healthcare, Chicago, IL). The unbound protein was purified by SEC using a Superdex 200 (GE Healthcare, Chicago, IL) equilibrated in PBS.
6.1. Evaluation of complex with huTNFα by size exclusion HPLC (SE-HPLC)
The complexes between anti-TNFα-ELP fusion mAb and huTNFα were characterized by SE-HPLC at room temperature on a Dionex Ultimate 3000 instrument (Dionex, Sunnyvale, CA) equipped with a Superose 6 10/300 GL column (GE Healthcare, Chicago, IL) with 1 × PBS, pH 7.4 as the mobile phase at a flow rate of 0.5 mL/min. Molar ratios of 1:1 huTNFα (trimer) to antibody were prepared and incubated at 37°C overnight with constant rotation at 350 rpm. 75 µL was then injected onto the SE-HPLC system and absorbance signals monitored at 280 nm.
6.2. Evaluation of hydrophobicity by hydrophobic chromatography (HIC)
Relative hydrophobicity of the anti-TNFα-ELP fusion mAb was characterized by HIC on a Dionex Ultimate 30,000 instrument (Dionex, Sunnyvale, CA) equipped with a TSK Gel Butyl NPR column (Tosoh, Japan) A linear gradient from 0 to 100% buffer B over 27 min was used to elute the protein at a flow rate of 1 mL/min. Mobile phase A consisted of 1.8-M ammonium sulfate, 20-mM phosphate, pH 7.2; whereas,mobile phase B had 20-mM phosphate and pH 7.2. Detection for the eluting proteins was performed by monitoring absorbance at 280 nm.
7. Evaluation of isoelectric point (pI)
The experimental pI of the protein material was determined using a cIEF immunoassay on a Peggy Sue instrument (Protein Simple, San Jose, CA). Briefly, samples were first diluted to 400 ng/mL in sample buffer. A premix was prepared by using ampholyte G2 premix pH 3–10 along with 4.4, 8.4, and 9.7 pI standards (Protein Simple, San Jose, CA) along with 1% TEMED. One volume of the sample was then mixed with three volumes of the premix. The proteins were then loaded into the capillaries and separated by charge. After separation, the proteins were immobilized onto the capillaries at their pI by exposure to UV light for 180 s. The detection antibody, goat Anti-Human IgG (H + L) HRP, cross-absorbed with bovine, horse, and mouse serum (Jackson Immunoresearch, West Grove, PA) diluted 1:200 was then flowed through the capillary and incubated for 60 min. Detection was done using luminol/peroxide chemiluminescence. Data obtained were then analyzed using the Compass software (Protein Simple, San Jose, CA).
7.1. Evaluation of thermal stability by differential scanning calorimetry (DSC)
Conformational stability analysis has been performed by using a MicroCal VP-DSC (Malvern, Westborough, MA) instrument. For the DSC measurements, samples were diluted to 0.5–1 mg/mL in PBS buffer pH 7.4. 420 µL of each sample was loaded into a 96 well plate and analyzed for changes in enthalpy associated with heat denaturation. All protein samples were subjected to a temperature ramp from 25°C to 90°C at a rate of 1°C/min and have been analyzed in single, n = 1. All datasets were averaged and standard deviations determined for each sample using specialized MicroCal thermal analysis software.
7.2. Evaluation of hydrodynamic radii by dynamic light scattering (DLS)
DLS temperature transitions studies were performed using two different instruments and methods. A high-throughput Wyatt DynaPro Plate Reader (Wyatt, Santa Barbara CA) equipped with an 830-nm laser source was used to determine the size distribution (Rh) profile when the fusion protein concentration was at 0.5 mg/mL in PBS. The temperature was steadily raised in increments of 0.065°C per minute. Sample was run in triplicates in a 384 well plate (Corning, Inc. NY), each well contained 50 µL. During the ramp, the protein solutions were measured successively, completing 10 5-s acquisitions for each sample well with auto-adjustment of attenuator and laser power. Averaged values between replicas have been recorded. The analysis and acquisition software used was Dynamics V7, Version 7.1.7 (Wyatt, Santa Barbara CA).
Size distribution was also collected on a Wyatt, DynaPro Nanostar (Wyatt, Santa Barbara). In this experiment, the anti-TNFα-ELP fusion mAb concentration was lowered to 0.1 mg/mL in PBS to control for solution viscosity and get a better estimate of particle size from the Stokes-Einstein equation. Light scattering data were collected at intervals of 2°C and samples were allowed to equilibrate and reach the new temperature before the next reading was recorded.
8. Pharmacokinetic evaluation
Studies were conducted in accordance with the AbbVie Institutional Animal Care and Use Committee guidelines. Groups of five male Sprague Dawley rats (Charles River Labs) were dosed intravenously with the anti-TNFα mAb at 5 mg/kg or the anti-TNFα-ELP at 8.33 mg/kg. Serial blood samples were collected from each animal for serum preparation at 1, 4, 24, 48, 72, 168, 240, 336, 504, and 672 h post dose. Concentration of anti-TNFα in each serum sample was quantified using a qualified MSD immunoassay. Briefly, biotinylated human TNFα was the capture reagent and a sulfo-tagged anti-human IgG was the detection reagent. Serum concentration over time data was fit and pharmacokinetic parameters estimated by non-compartmental analysis.
9. Analytical ultracentrifugation (AUC)
The concentration of the fusion protein was diluted to approximately 0.5 mg/mL in PBS, pH 7.4. The sample was then loaded into standard two-sector carbon epon centerpieces with a 1.2 cm optical path length, using sapphire windows. PBS at pH 7.4 was used for the reference buffer. The sample chamber and reference chamber were filled with 435 µL of sample and buffer, respectively. All samples were examined simultaneously using a 4-hole (AN-60Ti) rotor and a Beckman ProteomeLab XL-I analytical ultracentrifuge. Run conditions were programmed and centrifuge control was performed using ProteomeLab (v5.6). The samples and rotor were allowed to thermally equilibrate in the centrifuge for 2 h after the rotor temperature reached 20°±1°C. Sedimentation velocity conditions were performed at speed of 42,000 rpm, for 6 h. The numbers of scan acquired were 100 and the radial step size was 0.003 cm. The data were analyzed using the continuous S (c(s))-distribution analysis method as implemented by Peter Schuck in Sedfit v14.7g in order to understand the overall heterogeneity of the samples.
9.1. Evaluation of primary structure using mass spectrometry
Sample material was denatured with 8-M guanidine hydrochloride, 100-mM Tris pH 8.0 at a final guanidine hydrochloride concentration of 6 M. The denatured sample was then reduced with 10-mM Dithiothreitol (DTT) and alkylated with 25-mM iodoacetic acid at 37°C for 30 min. The entire reduced and alkylated samples were then desalted and exchanged into 100-mM Tris, pH 8.0 using Zeba Protein Desalt Spin Cartridges (Thermo Scientific, Waltham, MA). The desalted, reduced, and alkylated samples were then digested with sequencing grade trypsin (Roche, Indianapolis, IN) at a 1:20 w/w trypsin:protein ratio overnight at 37°C. The digestion was finally quenched by lowering the pH with formic acid. The digested samples were then separated on a 0.5 × 150 mm Jupiter 5-µm C18 column (Phenomenex, Torrance, CA) at a flow rate of 15 µL/min on a Waters Acquity LC system (Waters, Milford, MA) using a binary gradient (water:acetonitrile). The separated peptides were detected and identified via high-resolution mass spectrometry on a Velos Orbitrap MS (Thermo Scientific, Waltham, MA) using ESI MS/MS in positive ionization modes and CID fragmentation collision energy typically at 35 eV.
9.2. Neutralizing assay on TNFα induced cytotoxicity in mouse L929 cells
The murine fibrosarcoma cell line L929 (ATCC, #CCL-1 clone 929) was maintained by continuous passage in RPMI, 2-mM L-glutamine, 50 units/mL penicillin, 50 µg/mL streptomycin, 0.1-mM MEM non-essential amino and supplemented with 10% heat inactivated fetal bovine serum (Hyclone Labs, Logan, UT). Cells were split 1 day prior to assay. Next day, cells were plated at 5 × 104 cell/well on 96-well flat plates (Corning Costar, Corning, NY) and incubated for 2 h at 37°C, 5% CO2. During cell incubation, anti-TNFα-ELP fusion mAb and human IgG1 were serially diluted and pre-incubated with recombinant human TNFα (2.9 × 10−12 M final concentration) for 1 h at 25°C with gentle shaking. The pre-incubated samples were added to the cells and incubation was continued at 37°C, 5% CO2 for 20 h. To quantitate viability, WST-1 reagent (Roche, Indianapolis, IN) was added to wells according to manufacturer instructions. Plates were incubated under assay conditions for 3.5 h, centrifuged at 500 xg and supernatant transferred to an ELISA plate (Corning Costar, Corning, NY). The plates were read at OD 420–600 nm on a Spectramax 190 ELISA plate reader (Molecular Devices, Sunnyvale, CA). The IC50 value was determined by plotting the OD420–600 values vs. the concentration of mAb-ELP using a non-linear 4 parameter curve fit using Graphpad Prism 5 (Graphpad, La Jolla, CA).
9.3. Internalization assay using stably transfected cell line HEK293H-tmTNF
HEK293H cells were stably transfected with human TNFα. They express the transmembrane form on their cell surface. The transfected cells were harvested, washed with D-PBS, and resuspended at 1E4 cell/mL in assay media: DMEM media (Gibco, Grand Island, NY), 50 units/mL penicillin/50 µg/mL streptomycin, 0.1 mM MEM non-essential amino acids, sodium pyruvate, and supplemented with 10% fetal bovine serum (Hyclone labs, Logan, UT). Cells were plated at 1E3 cells/well on 96-well black plates (Corning Costar, Corning, NY) and incubated for 18 h at 37°C, 5% CO2. Following incubation, cells were washed with ice-cold assay buffer (3% FBS in D-PBS) and placed on ice. Test articles were serially diluted in ice-cold assay buffer and added to the cells on ice for 1 h. Cells were washed twice with ice-cold assay buffer and centrifuged for 5 min at 350 × g, 4°C. An anti-human IgG Fab conjugated to saporin (Advanced Targeting Systems, San Diego) was diluted in ice-cold assay buffer and added to the cells for 1 h on ice. Following incubation, cells were washed twice to remove free secondary antibody. Assay media was added to wells and plates were incubated for 72 h at 37°C, 5% CO2. To quantitate viability, Cell Titer Glo (Promega, Madison, WI) was added to wells according to manufacturer instructions. Luminescence was read using a Topcount (Perkin Elmer, Waltham, MA). The IC-50 value was determined by plotting the RLU vs. the concentration of mAb-ELP using a non-linear four parameter curve fit using Graphpad Prism 5 (Graphpad, La Jolla, CA).
9.4. Sample preparation for electron microscopy
Imaging of anti-TNFα-ELP fusion mAb and binding to antigen was performed at NanoImaging Services Inc as a fee for service. Details of the method used in a previous study with NanoImaging Services Inc are described in Correia, Sung et al., 2013 (Correia et al., Citation2013). In brief, anti-TNFα-ELP fusion mAb, supplied at 0.2 mg/mL, was diluted 20 fold in 20-mM HEPES buffer prior to imaging. The samples were imaged on a layer of continuous carbon supported by nitro-cellulose on a 400-mesh copper grid. The grids were prepared by applying 3 μL of sample suspension to a cleaned grid, blotting away with filter paper, and immediately staining with uranyl formate. Electron microscopy was performed using an FEI Tecnai T12 electron microscope, operating at 120 keV equipped with an FEI Eagle 4 K × 4 K CCD camera. Negative stain grids were transferred into the electron microscope using a room-temperature stage. Images were acquired at multiple scales using the automated image acquisition software package Leginon (Suloway et al., Citation2005). After identifying potentially suitable target areas for imaging at lower magnifications, high-magnification images were acquired at nominal magnifications of 110,000× (0.10 nm/pixel), 67,000× (0.16 nm/pixel), and 52,000× (0.21 nm/pixel). The images were acquired at a nominal under focus of −3 μm to −2 μm and electron doses of ~25–35 e- Å2.
9.5. Image processing
Image processing was performed using the Appion software package (Lander et al., Citation2009). Contrast transfer functions of the images were corrected using Ace2 (Mallick, Carragher, Potter, & Kriegman, Citation2005) and CTFFind v3 (Mindell & Grigorieff, Citation2003). Individual particles in the 67,000× images were selected using automated picking protocols, followed by several rounds of reference-free alignment and classification based on the XMIPP (Sorzano et al., Citation2004) processing package to sort them into self-similar groups. A minimum of 200 particles were assembled to create a class average.
9.6. Biacore binding to human TNFα
Binding of anti-TNFα antibodies to recombinant human TNFα was assessed with the use of Biacore T200 (GE Healthcare) instrument. S series CM 5 chip with goat anti-human IgG Fc antibodies that were directly immobilized via amino coupling on all four flow cells to the density of approximately 10,000 RU according to GE protocol was used for experiments. Antibodies, diluted to a concentration of ~ 0.5 − 1.0 µg/mL in HBS-EP+ buffer, were captured on flow cells 2, 3, and 4 at a flow rate of 5 µL/min. Flow cell 1 was used as a reference surface. HBS-EP+, was used as a running buffer. Recombinant human TNFα (0, 57.6, 28.8, 14.4, 7.2, 3.6, 1.8 nM) was injected over all flow cells for 5 min at a flow rate of 50 µL/min to ascertain association rates. Association phase of the experiment was immediately followed by a 15 min dissociation phase that consisted of continuous flow of the running buffer at a flow rate of 50 µL/min. Chip surfaces were regenerated with two consecutive pulses (20 s and 10 s) of 10 mM Glycine, pH 1.5, administered at 100 µL/min after each experimental cycle. Each concentration point was run in duplicate.
Biacore T200 Evaluation Software version 2.0 was used to fit the data from the TNF binding studies to a 1:1 kinetic model. Duplicate experimental values were simultaneously fitted by the software to produce single value for on and off kinetic rates and overall affinity for each of the samples.
Abbreviations
| cIEF | = | Capillary Isoelectric focusing |
| DSC | = | Differential Scanning Calorimetry |
| ECM | = | Extracellular Matrix |
| ELP | = | Elastin like polypeptides |
| Fab | = | Fragment antigen binding |
| Fc | = | Fragment crystallizable |
| GI | = | Gastro-intestinal |
| IA | = | Inter-articular |
| IgG | = | Immunoglobulin |
| IL-1RA | = | Interleukin-1 receptor antagonist |
| MHC | = | Major Histocompatibility Complex |
| OA | = | Osteoarthritis |
| PD | = | Pharmacodynamics |
| PK | = | Pharmacokinetics |
| PMMA | = | Polymethylmethacrylate |
| RA | = | Rheumatoid arthritis |
| Rh | = | Hydrodynamic radius |
| SpA | = | Spondyloarthritis |
| SPR | = | Surface Plasmon Resonance |
| sTNFRII | = | Soluble Tumor Necrosis Factor Receptor II |
| TACE | = | TNF alpha converting enzyme |
| TEM | = | Transmission Electron Microscopy |
| TNFα | = | Tumor Necrosis Factor Alpha |
AbbVie disclosures
The design, study conduct, and financial support for the study were provided by AbbVie (formerly Abbott). AbbVie participated in the interpretation of data, review, and approval of the publication; all authors contributed to the development of the publication and maintained control over the final content.
Internal Author disclosures
Ramkrishna Sadhukhan, Nathan Brown, David Ouellette, Dana I. Filoti, David Winarta, David Banach, Reema Raghavendra, Silvino Sousa, Leslie Alessandri, Anhdao Darcy, Sahana Bose, Lucia Eaton, Gregory Preston, and Ivan Correia are AbbVie employees and may hold AbbVie stock and/or options.
Cover image
Source: Jennifer DiMauro.

Supplemental Material
Download Zip (346.5 KB)Acknowledgements
The authors are grateful to Wolfgang Fraunhofer, Li Zhou, Trudi Veldman, and Jochen Salfeld, for feedback, guidance and support for this study.
Supplementary material
Supplementary material for this article can be accessed here https://doi.org/10.1080/23312025.2018.1469387
Additional information
Funding
Notes on contributors

Ramkrishna Sadhukhan
As Head of the Protein Engineering and Expression (PE&E) group; my team helps in design and expression of conventional as well as novel biologics therapeutics. Early in discovery we evaluate different formats, drug like properties as well as activity of engineered molecules. Upon selecting a lead molecule(s) we work with other cross functional teams to advance development; involving scale-up, purification and characterization activities (Protein Sciences colleagues), ensuring optimal affinity (Biologics Generation colleagues), in vitro activity (Immunology colleagues), early formulation activity (Pre-formulation colleagues) and pharmacokinetics (Drug Metabolism and Pharmacokinetic colleagues). Engineering unique properties (thermo-responsiveness) into therapeutic proteins provide new opportunities for disease targeting and treatment options. It takes a large team of scientists with different skill sets to advance a clinical candidate.
Related Research Data
References
- Baker, K., Qiao, S.-W., Kuo, T. T., Aveson, V. G., Platzer, B., Andersen, J.-T., … Blumberg, R. S. (2011). Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proceedings of the National Academy of Sciences of the United States of America, 108, 9927–19. doi:10.1073/pnas.1019037108
- Betre, H., Liu, W., Zalutsky, M. R., Chilkoti, A., Kraus, V. B., & Setton, L. A. (2006). A thermally responsive biopolymer for intra-articular drug delivery. Journal of Controlled Release: Official Journal of the Controlled Release Society, 115, 175–182. doi:10.1016/j.jconrel.2006.07.022
- Blanco, R., Martínez-Taboada, V. M., Villa, I., González-Vela, M. C., Fernández-Llaca, H., Agudo, M., & González-López, M. A. (2009). Long-term successful adalimumab therapy in severe hidradenitis suppurativa. Arch Dermatol, 145, 580–584. doi:10.1001/archdermatol.2009.49
- Brehm, M. A., Daniels, K. A., & Welsh, R. M. (2005). Rapid production of TNF-following TCR engagement of naive CD8 T-cells. The Journal of Immunology, 175, 5043–5049. doi:10.4049/jimmunol.175.8.5043
- Cawthorn, W. P., & Sethi, J. K. (2008). TNF-α and adipocyte biology. FEBS Letters, 582, 117–131. doi:10.1016/j.febslet.2007.11.051
- Conrad, U., Plagmann, I., Malchow, S., Sack, M., Floss, D. M., Kruglov, A. A., … Scheller, J. (2011). ELPylated anti-human TNF therapeutic single-domain antibodies for prevention of lethal septic shock. Plant Biotechnology Journal, 9, 22–31. doi:10.1111/pbi.2010.9.issue-1
- Correia, I., Sung, J., Burton, R., Jakob, C. G., Carragher, B., Ghayur, T., & Radziejewski, C. (2013). The structure of dual-variable-domain immunoglobulin molecules alone and bound to antigen. mAbs, 5, 364–372. doi:10.4161/mabs.24258
- de Waal Malefyt, R., Abrams, J., Bennett, B., Figdor, C. G., & de Vries, J. E. (1991). Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. The Journal of Experimental Medicine, 174, 1209–1220. doi:10.1084/jem.174.5.1209
- Evans, C. H., Kraus, V. B., & Setton, L. A. (2014). Progress in intra-articular therapy. Nature reviews. Rheumatology, 10, 11–22.
- Fiocco, U., Sfriso, P., Oliviero, F., Roux-Lombard, P., Scagliori, E., Cozzi, L., … Punzi, L. (2010). Synovial effusion and synovial fluid biomarkers in psoriatic arthritis to assess intraarticular tumor necrosis factor-α blockade in the knee joint. Arthritis Research & Therapy, 12, R148. doi:10.1186/ar3090
- Fioravanti, A., Fabbroni, M., Cerase, A., & Galeazzi, M. (2009). Treatment of erosive osteoarthritis of the hands by intra-articular infliximab injections: A pilot study. Rheumatology International, 29, 961–965. doi:10.1007/s00296-009-0872-0
- Fisher, B. A., & Keat, A. (2006). Should we be using intraarticular tumor necrosis factor blockade in inflammatory monoarthritis? The Journal of Rheumatology, 33, 1934–1935.
- Havell, E. A., & Rogerson, B. J. (1993). Endotoxin-induced tumor necrosis factor alpha synthesis in murine embryo fibroblasts. Infection and Immunity, 61, 1630–1635.
- Ho, L.-J., Wang, -J.-J., Shaio, M.-F., Kao, C.-L., Chang, D.-M., Han, S.-W., & Lai, J.-H. (2001). Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. The Journal of Immunology, 166, 1499–1506. doi:10.4049/jimmunol.166.3.1499
- Hollander, J. L., Brown, E. M., Jr., Jessar, R. A., & Brown, C. Y. (1951). Hydrocortisone and cortisone injected into arthritic joints; comparative effects of and use of hydrocortisone as a local antiarthritic agent. Journal of the American Medical Association, 147, 1629–1635. doi:10.1001/jama.1951.03670340019005
- Imaizumi, T., Itaya, H., Fujita, K., Kudoh, D., Kudoh, S., Mori, K., … Satoh, K. (2000). Expression of tumor necrosis factor-alpha in cultured human endothelial cells stimulated with lipopolysaccharide or interleukin-1alpha. Arteriosclerosis, Thrombosis, and Vascular Biology, 20, 410–415. doi:10.1161/01.ATV.20.2.410
- Ionescu, R. M., Vlasak, J., Price, C., & Kirchmeier, M. (2008). Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. Journal of Pharmaceutical Sciences, 97, 1414–1426. doi:10.1002/jps.21104
- Kowalczyk, T., Hnatuszko-Konka, K., Gerszberg, A., & Kononowicz, A. K. (2014). Elastin-like polypeptides as a promising family of genetically-engineered protein based polymers. World Journal of Microbiology & Biotechnology, 30, 2141–2152. doi:10.1007/s11274-014-1649-5
- Kriegler, M., Perez, C., DeFay, K., Albert, I., & Lu, S. D. (1988). A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell, 53, 45–53. doi:10.1016/0092-8674(88)90486-2
- Lander, G. C., Stagg, S. M., Voss, N. R., Cheng, A., Fellmann, D., Pulokas, J., … Carragher, B. (2009). Appion: An integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology, 166, 95–102. doi:10.1016/j.jsb.2009.01.002
- Langer, R., & Tirrell, D. A. (2004). Designing materials for biology and medicine. Nature, 428, 487–492. doi:10.1038/nature02388
- Luginbuhl, K. M., Schaal, J. L., Umstead, B., Schaal, J. L., Umstead, B., Mastria, E. M., … Chilkoti, A. (2017). One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nature Biomedical Engineering, 1, 78. doi:10.1038/s41551-017-0078
- MacEwan, S. R., & Chilkoti, A. (2010). Elastin-like polypeptides: Biomedical applications of tunable biopolymers. Biopolymers, 94, 60–77. doi:10.1002/bip.21327
- Mallick, S. P., Carragher, B., Potter, C. S., & Kriegman, D. J. (2005). ACE: Automated CTF estimation. Ultramicroscopy, 104, 8–29. doi:10.1016/j.ultramic.2005.02.004
- Marshall, K. W. (2000). Intra-articular hyaluronan therapy. Current Opinion in Rheumatology, 12, 468–474. doi:10.1097/00002281-200009000-00022
- McMillan, R. A., Caran, K. L., Apkarian, R. P., & Conticello, V. P. (1999). High resolution topographic imaging of environmentally responsive elastin-mimetic hydrogels. Macromolecules, 32, 9067–9070. doi:10.1021/ma991119z
- McMillan, R. A., & Conticello, V. P. (2000). Synthesis and characterization of elastin-mimetic protein gels derived from a well defined polypeptide precursor. Macromolecules, 33, 4809–4821. doi:10.1021/ma9921091
- Mindell, J. A., & Grigorieff, N. (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology, 142, 334–347. doi:10.1016/S1047-8477(03)00069-8
- Nagapudi, K., Brinkman, W., Leisen, J. E., Huang, L., McMillan, R. A., Apkarian, R. P., … Chaikof, E. L. (2002). Photomediated solid-state cross-linking of an elastin−mimetic recombinant protein polymer. Macromolecules, 35, 1730–1737. doi:10.1021/ma011429t
- Nakao, A., Fukushima, H., Kajiya, H., Ozeki, S., & Okabe, K. (2007). RANKL-stimulated TNFα production in osteoclast precursor cells promotes osteoclastogenesis by modulating RANK signaling pathways. Biochemical and Biophysical Research Communications, 357, 945–950. doi:10.1016/j.bbrc.2007.04.058
- Peppas, N. A., & Langer, R. (1994). New challenges in biomaterials. Science, 263, 1715–1720. doi:10.1126/science.8134835
- Perez, C., Albert, I., DeFay, K., Zachariades, N., Gooding, L., & Kriegler, M. (1990). A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell, 63, 251–258. doi:10.1016/0092-8674(90)90158-B
- Rath, T., Baker, K., Pyzik, M., & Blumberg, R. S. (2014). Regulation of immune responses by the neonatal fc receptor and its therapeutic implications. Frontiers in Immunology, 5, 664.
- Rath, T., Kuo, T. T., Baker, K., Qiao, S.-W., Kobayashi, K., Yoshida, M., … Blumberg, R. S. (2013). The immunologic functions of the neonatal Fc receptor for IgG. Journal of Clinical Immunology, 33(Suppl 1), S9–S17. doi:10.1007/s10875-012-9768-y
- Sakellariou, G. T., Kakavouli, G., & Chatzigiannis, I. (2006). Intraarticular injection of infliximab. The Journal of Rheumatology, 33, 1912–1913. author reply 1913.
- Shamji, M. F., Betre, H., Kraus, V. B., Chen, J., Chilkoti, A., Pichika, R., … Setton, L. A. (2007). Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: Sustained release of a local antiinflammatory therapeutic. Arthritis & Rheumatism, 56, 3650–3661. doi:10.1002/(ISSN)1529-0131
- Shamji, M. F., Chen, J., Friedman, A. H., Richardson, W. J., Chilkoti, A., & Setton, L. A. (2008). Synthesis and characterization of a thermally-responsive tumor necrosis factor antagonist. Journal of Controlled Release: Official Journal of the Controlled Release Society, 129, 179–186. doi:10.1016/j.jconrel.2008.04.021
- Shields, R. L., Namenuk, A. K., Hong, K., Meng, Y. G., Rae, J., Briggs, J., … Presta, L. G. (2001). High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. The Journal of Biological Chemistry, 276, 6591–6604. doi:10.1074/jbc.M009483200
- Simkin, P. A. (1995). Synovial perfusion and synovial fluid solutes. Annals of the Rheumatic Diseases, 54, 424–428. doi:10.1136/ard.54.5.424
- Simkin, P. A., Bassett, J. E., & Koh, E. M. (1995). Synovial perfusion in the human knee: A methodologic analysis. Seminars in arthritis and rheumatism, 25, 56–66.
- Sorzano, C. O., Marabini, R., Velázquez-Muriel, J., Bilbao-Castro, J. R., Scheres, S. H. W., Carazo, J. M., & Pascual-Montano, A. (2004). XMIPP: A new generation of an open-source image processing package for electron microscopy. Journal of Structural Biology, 148, 194–204. doi:10.1016/j.jsb.2004.06.006
- Suloway, C., Pulokas, J., Fellmann, D., Cheng, A., Guerra, F., Quispe, J., … Carragher, B. (2005). Automated molecular microscopy: The new Leginon system. Journal of Structural Biology, 151, 41–60. doi:10.1016/j.jsb.2005.03.010
- Tatham, A. S., & Shewry, P. R. (2000). Elastomeric proteins: Biological roles, structures and mechanisms. Trends in Biochemical Sciences, 25, 567–571. doi:10.1016/S0968-0004(00)01670-4
- Urry, D. W., Parker, T. M., Reid, M. C., & Gowda, D. C. (1991). Biocompatibility of the bioelastic materials, poly(GVGVP) and its γ-Irradiation cross-linked matrix: Summary of generic biological test results. Journal of Bioactive and Compatible Polymers, 6, 263–282. doi:10.1177/088391159100600306
- van der Zee, H. H., Laman, J. D., de Ruiter, L., Dik, W. A., & Prens, E. P. (2012). Adalimumab (antitumour necrosis factor-α) treatment of hidradenitis suppurativa ameliorates skin inflammation: An in situ and ex vivo study. British Journal of Dermatology, 166, 298–305. doi:10.1111/j.1365-2133.2011.10698.x
- Wajant, H., Pfizenmaier, K., & Scheurich, P. (2003). Tumor necrosis factor signaling. Cell Death and Differentiation, 10, 45–65. doi:10.1038/sj.cdd.4401189
- Williamson, B. D., Carswell, E. A., Rubin, B. Y., Prendergast, J. S., & Old, L. J. (1983). Human tumor necrosis factor produced by human B-cell lines: Synergistic cytotoxic interaction with human interferon. Proceedings of the National Academy of Sciences of the United States of America, 80, 5397–5401. doi:10.1073/pnas.80.17.5397