
ABSTRACT
Design of specific kinase inhibitors is an appealing approach for developing new anticancer treatments. However, only a few success stories have been reported to date. Here we demonstrate how the combination of old-fashioned and new biophysical tools together with recent advances in genomics and molecular evolution can aid in overcoming existing limitations.
We get cancer because we are multicellular
Multicellularity is fundamentally based on successful communication between cells, and protein kinase-mediated protein phosphorylation has played a key role in the development of coordinated signal transduction between cells.Citation1 Protein kinases themselves are molecular switches as they readily interconvert between the active and inactive state through a multitude of regulation mechanisms.Citation2 However, just like switches constructed by humans, life's switches can go wrong (i.e., mutations) and when they do, cells stop playing by the rules. Typically, mutations that result in an overactive protein kinase will cause the signal transduction pathway to remain turned on, which often leads to cancer. Thus protein kinases allow for coordination between cells, and therefore complexity, but are often also the cause of cancer due to misregulation.
The story of 2 cancer-associated tyrosine kinases
The detrimental role of kinases in cancer has made them a leading cancer drug target for the pharmaceutical industry, but designing such therapeutics is a daunting challenge with only limited successful outcomes. A prominent example of such success is the anticancer drug Gleevec, a well-known treatment for chronic myeloid leukemia.Citation3 The reason for Gleevec's impressive success lies in its incredible selectivity for human Abelson murine leukemia viral oncogene homolog 1 (ABL1) kinase domain over its closest homologs, the sarcoma proto-oncogene (SRC) subfamily of kinases. Gleevec was the first drug approved by the U.S. Food and Drug Administration (FDA) that specifically targeted the kinase of interest and is often touted as a proof of principle for the feasibility of rationally designed drugs. However, the mechanism behind its remarkable selectivity remained elusive for decades.
In our recent work,Citation4 by combining nuclear magnetic resonance spectroscopy (NMR) and old-fashioned kinetics of stopped-flow fluorescence spectroscopy we showed that selectivity is rooted in the differential energy landscapes, and not single static structures. The kinases differ in their equilibrium between conformational substates of the kinase-drug complex, and this is the reason for Gleevec's success as a cancer therapeutic. This is in contrast to the popular hypothesis within the field proposing conformational selection by the DFG-loop, a fully conserved loop in the kinase kingdom. Importantly, long-range interactions are responsible for the vast differences in drug affinity among the kinases, highlighting the importance of residues away from the immediate binding pocket. However, the atomistic players responsible for the observed change in kinase-Gleevec dynamics remained unknown.Citation4
Sequence comparison?
As the central dogma teaches us, genetic information is encoded in DNA and then transferred to the protein, thus determining its properties. Thus, to ultimately pinpoint the differences “that matter” one needs to compare sequences of a tight binder (e.g., ABL1) and a weak binder (e.g., SRC). Unfortunately reality is always more complicated than a thought experiment. Modern proteins have gone through many rounds of random mutations and natural selection and therefore carry a large number of differences. For example, the catalytic domains of SRC and ABL1 differ by approximately 150 amino acids, most of which are irrelevant for Gleevec binding. Proteins have an evolutionary history, a maze through which they have moved that ends in the differential function we observe today.Citation5
It is becoming more and more apparent that differentiation of function is not simply the result of single, double, or even triple point mutations but rather the result of changes in networks of residues interacting with each other, some far from the site of action.Citation6 Identifying the residues involved in these networks is very difficult and often impossible by simple comparison of modern day sequences.
If life closes a door, it opens a window
Fortunately there is a way around these problems that are created by evolution, and ironically it is evolutionary-based. Very early on, Linus Pauling and Emile Zuckerkandl recognized that through sequence comparison from a variety of organisms the phylogeny relating the observed protein sequences could be inferred.Citation7 Furthermore, these authors realized that with enough sequences it would be possible to “reconstruct” sequences at the root of these inferred phylogenies. These reconstructed proteins would have existed in the now extinct common ancestors of the modern day organisms making up the phylogeny.
Modern techniques of gene synthesis allow us to recreate these extinct proteins in the laboratory and to use them as a tool to understand the differences between modern day proteins. This ancestral sequence reconstruction (ASR) approach allows for the recreation of the sequence background in which the functional switch occurred and, additionally, simplifies the problem by reducing the number of residues suspected to be involved in the observed functional switch.Citation8
Unravelling a modern cancer durg's mechanism using ancient proteins
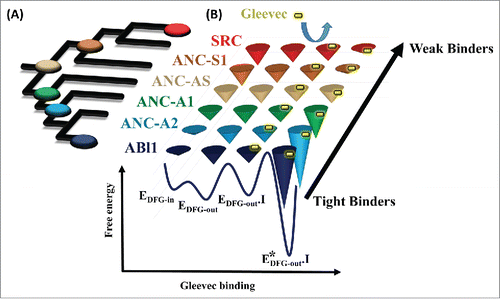
The power of ASR, when combined with traditional biochemical and biophysical techniques, was highlighted in a recent paper published by our laboratory,Citation9 which finally revealed the molecular players responsible for the difference in Gleevec's affinity to SRC and ABL1. We began by calculating a detailed phylogeny using protein kinase sequences spanning the entire metazoan kingdom that crucially included sequences from the class of Choanoflagellates, which are thought to be the single cell ancestors of the metazoan lineage.Citation10 The resurrected common ancestor between Choanoflagellates and H. sapiens, estimated to have existed a little over 1 billion years ago, had an intermediate affinity for Gleevec. By walking along the phylogenetic branches toward SRC and ABL1 and using a combination of pre-steady state kinetics of drug binding, x-ray crystallography, and sequence comparison, we were able to piece together an answer to what differed between the kinases that weakly or tightly bound Gleevec (see ). These experiments identified 15 residues that turned a weak Gleevec binder into a strong one. Because of epistasis and extensive sequence divergence such analysis would have not been possible if only the modern enzymes were considered.
Figure 1. Evolution of differential energy landscapes of Gleevec binding to SRC, ABL1, and reconstructed ancestors. (A) Phylogenetic tree of the SRC/ABL subfamilies of tyrosine kinases, with reconstructed nodes shown as colored buttons. (B) Schematic representation of the evolution of the Gleevec binding free energy landscape. Depth of the conical wells represents the free energy of a state. The barriers between the states represent the frequencies of interconversion between the states. Gleevec in a free state and bound to the kinases is illustrated as a yellow pill.
Detailed understanding of the molecular mechanism of Gleevec's selectivity opens doors for new routes in rational drug design. As new data indicate, design efforts need to go beyond optimizing drug interactions with the binding pocket. Instead, the dynamics of kinases and kinase–drug complexes need to be considered. Identifying dynamic networks in targets and ultimately using them in drug design is a challenging task. However, by extracting billions of bits of information encoded in protein sequence evolution and putting them to good use, ASR can be a powerful addition to the existing experimental and computational methods to help decipher such dynamic networks. The era of sequencing and genomics has made it possible to unravel the evolutionary history of clinically relevant targets, which can and should be utilized in drug development efforts.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 2009; 21(2):140-6; PMID:19269802; http://dx.doi.org/10.1016/j.ceb.2009.01.028.
- Rokas A. The molecular origins of multicellular transitions. Curr Opin Genet Dev 2008; 18(6):472-8; PMID:18926910; http://dx.doi.org/10.1016/j.gde.2008.09.004.
- Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res 2002; 8(5):935-42; PMID:12006504.
- Agafonov RV, Wilson C, Otten R, Buosi V, Kern D. Energetic dissection of Gleevec's selectivity toward human tyrosine kinases. Nat Struct Mol Biol 2014; 21(10):848-53; PMID:25218445; http://dx.doi.org/10.1038/nsmb.2891.
- DePristo M.A, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat Rev Genet 2005; 6(9):678-87; PMID:16074985; http://dx.doi.org/10.1038/nrg1672.
- Vishveshwara S, Ghosh A, Hansia P. Intra and inter-molecular communications through protein structure network. Curr Protein Pept Sci 2009; 10(2):146-60; PMID:19355982; http://dx.doi.org/10.2174/138920309787847590.
- Pauling L and Zuckerkandl E, Chemical Paleogenetics: Molecular ‘Restoration Studies’ of Extinct Forms of Life, Acta Chem. Scand. 1963; 17:S9–S16.
- Harms MJ, Thornton JW. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet 2013; 14(8):559-71; PMID:23864121; http://dx.doi.org/10.1038/nrg3540.
- Wilson C, Agafonov RV, Hoemberger M, Kutter S, Zorba A, Halpin J, Buosi V, Otten R, Waterman D, Theobald DL, et al. Kinase dynamics. Using ancient protein kinases to unravel a modern cancer drug's mechanism. Science 2015; 347(6224):882-6; PMID:25700521; http://dx.doi.org/10.1126/science.aaa1823.
- King N. The unicellular ancestry of animal development. Dev Cell 2004; 7(3):313-25; PMID:15363407; http://dx.doi.org/10.1016/j.devcel.2004.08.010.