
ABSTRACT
Sequencing studies have been instrumental in understanding the genetic basis of chronic lymphocytic leukemia (CLL). Our recent whole-genome sequencing study focusing on lower cytogenetic risk CLL demonstrated that CLL mutations can be attributed to 3 key mutational processes—2 types of activation induced-cytidine deaminase (AID) signatures and an aging signature—that operate at different times throughout CLL evolution.
Chronic lymphocytic leukemia (CLL) is a common incurable malignancy of CD5+ B lymphocytes that is clinically extremely heterogeneous. In recent years, sequencing projects undertaken by our group and others have identified key mutations underlying the marked biologic heterogeneity of this disease. Two seminal papers by Landau et al.Citation1 and Quesada et al.Citation2 identified 20 significantly mutated genes in CLL. However, these studies had low power to detect infrequently mutated genes, leading to larger follow-up studies that have identified driver genes mutated at lower frequencies.Citation3,4 Interestingly, most of these candidate driver gene mutations have been consistently associated with higher risk CLL, in particular with unmutated immunoglobulin heavy chain variable region (IGHV) genes and higher risk FISH abnormalities (TP53 [best known as p53] with 17p; SF3B1 with 11q; NOTCH1 with +12 and 17p). In fact, each of these recent large studies, which looked at 538 and 506 (including 54 cases of monoclonal B-cell lymphocytosis [MBL]) patients, respectively,Citation3,4 identified a percentage of CLLs (approximately 25% and 40%) with no apparent driver event, or with isolated deletion of 13q14 as the sole driver event. In mice, the latter event results in a spectrum of B-cell malignancies (some CLL-like), but with low penetrance,Citation5 and therefore is probably insufficient to cause CLL in humans.
Thus, the genetic drivers of lower cytogenetic risk CLL, which represents >50% of cases, are relatively unknown, and this gap formed the basis of our recent study published in Nature Communications.Citation6 We performed whole-genome sequencing on a CLL cohort (n=30) strongly enriched in patients with del(13q) or normal cytogenetics while controlling for their IGHV mutational status. Analysis of the distribution of non-silent coding mutations in important genesets revealed the presence of 3 mutually exclusive subgroups: (1) 17% were cases with mutations in immunoglobulin lambda-like polypeptide 5 (IGLL5), a previously undescribed recurrently mutated gene in CLL; (2) 33% had mutations in at least one of the 20 CLL driver genes enriched in normal cytogenetics and unmutated IGHV;Citation1 and (3) 23% with no obvious mutations of interest, that were enriched in del(13q).
Interestingly, we noted that the pattern of mutations in the IGLL5 gene was strongly suggestive of activation induced-cytidine deaminase (AID) activity, which was not identified as a global mutational mechanism in CLL in earlier efforts through unsupervised genome-wide analysis. In fact, canonical AID activity had been reported in CLL using only supervised analysis and was thought to be predominantly confined to the immunoglobulin loci and a few off-target sites. Our collaborator, Jaegil Kim from the Getz group at the Broad Institute, therefore developed a modified Bayesian non-negative matrix factorization algorithm (BayesNMF) to infer the presence of mutational signatures in our dataset and their sample-specific contributions.Citation6 This approach is similar to that recently reported by Alexandrov et al.,Citation7 except that we included mutational distance as an additional factor because we noted that mutations at AID-type target sites showed clustering in our dataset.
Our analysis was able to identify canonical AID (c-AID) activity as a global signature in CLL (). We also identified a non-canonical AID (nc-AID) activity that accounted for 20% of mutationsCitation6 (). These two signatures arise from differential repair mechanisms after DNA lesions are induced by AID. Specifically, the c-AID signature arises from either direct replication over AID-induced U:G lesions or removal of the uracil by uracil DNA glycosylase (UNG) followed by replication. Alternatively, nc-AID-related mutations result from processing of the AID-induced lesions by the error prone DNA polymerase η, part of the mismatch repair pathway. Together, these 2 mechanisms accounted for 25% of mutations, while an aging signature accounted for approximately 75% of mutations. Interestingly, 95% of observed coding region mutations were associated with the aging signature, which is therefore likely to be the primary contributor to coding driver mutations in CLL. Around the same time, Puente et al. also reported the genome-wide activity of these 3 signatures in their cohort,Citation4 further validating our findings.
Figure 1. Discovery of mutational signatures in chronic lymphocytic leukemia. The catalog of mutations from 30 CLL whole genomes was deconvoluted into 3 distinct mutational signatures—aging, canonical activation induced-cytidine deaminase (c-AID), and non-canonical AID (nc-AID)—using a modified non-negative matrix factorization (NMF) algorithm that accounts for the distance between mutations.
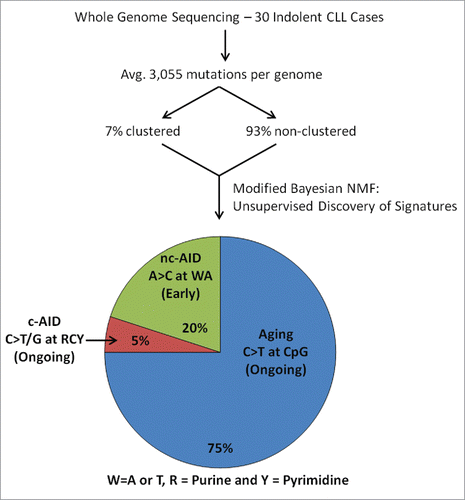
As in our prior work,Citation1 we analyzed the clonality of each mutation in our dataset to estimate its time of onset. Clonal mutations happen earliest in the lifetime of the tumor, and smaller subclones arise subsequently. Based on the distribution of newer subclonal and older clonal mutations within each signature, we proposed that nc-AID-induced mutations mostly occur at earlier times whereas the c-AID and aging mechanisms continue to operate within the CLL cell. Although it is not surprising that the aging mechanism is ongoing, it is of interest that we were able to detect more recent subclonal mutations due to c-AID activity given that AID expression is at best minimal in circulating CLL cells. The Chiorazzi and Dighiero groups have shown that AID expression is restricted to proliferating cells, which are sparse in CLL and typically present in tissues at the time of proliferation, and to patients with higher risk unmutated IGHV.Citation8,9 Indeed, we found that although the overall c-AID activity is higher in mutated IGHV cases, as expected, the ongoing c-AID activity is enriched in higher risk unmutated IGHV.
How this ongoing c-AID activity influences the mutational status of IGHV and other genes, as well as disease outcome in the long term, is an interesting unanswered question. Preliminary findings from double transgenic AID/Eµ-TCL1 mice indicate a more aggressive disease and shorter overall survival with constitutive increase in AID expression,Citation10 raising the question of whether AID might contribute to ongoing genomic instability.
In summary, a comprehensive catalog of mutations in CLL has now been established by large-scale sequencing studies. Together, these papers provide compelling evidence for the association of several recurrently mutated CLL genes and disease outcome. Using whole-genome sequencing, our paper by Kasar et al. has delved deeper into the underlying mutational processes that contribute to the development of these mutations throughout tumor evolution. By incorporating the distance between mutations into our modified Bayesian NMF algorithm, we were able to discern a distinct genome-wide c-AID signal in an unsupervised manner and establish that it can be ongoing in tumor evolution. Our algorithm can also be broadly applied to the discovery of clustering dependent mutational processes in other cancers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
SK and JRB wrote the manuscript. JRB directed the study, wrote, and reviewed the manuscript.
Acknowledgements
We thank all our co-authors from the Kasar et al. paper (Nat Comm 2015) paper, especially Drs. Gad Getz and Jaegil Kim. The signature discovery algorithm described here was developed by Dr. Kim under the guidance of Dr. Getz (Paul C. Zamecnik, MD, Chair in Oncology at Massachusetts General Hospital).
Funding
This work was funded by the Melton Family Fund for CLL Research and the National Human Genome Research Institute (NHGRI) Grant U54HG003067. JRB is a clinical scholar of the Leukemia Lymphoma Society and is supported by the American Cancer Society (RSG-13-002-01-CCE) and the Leukemia Lymphoma Society (TRP#6289-13).
References
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152, 714-26; PMID:23415222; http://dx.doi.org/10.1016/j.cell.2013.01.019
- Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2012; 44, 47-52; PMID:22158541; http://dx.doi.org/10.1038/ng.1032
- Landau DA, et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015; 526, 525-30; PMID:26466571; http://dx.doi.org/10.1038/nature15395
- Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, Munar M, Rubio-Pérez C, Jares P, Aymerich M, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015; 526, 519-24; PMID:26200345; http://dx.doi.org/10.1038/nature14666
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010; 17, 28-40; PMID:20060366; http://dx.doi.org/10.1016/j.ccr.2009.11.019
- Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, Lawrence MS, Kiezun A, Fernandes SM, Bahl S, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun 2015; 6, 8866; PMID:26638776; http://dx.doi.org/10.1038/ncomms9866
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature 2013; 500, 415-21; PMID:23945592; http://dx.doi.org/10.1038/nature12477
- Oppezzo P, Vuillier F, Vasconcelos Y, Dumas G, Magnac C, Payelle-Brogard B, Pritsch O, Dighiero G. Chronic lymphocytic leukemia B cells expressing AID display dissociation between class switch recombination and somatic hypermutation. Blood 2003; 101, 4029-32; PMID:12521993; http://dx.doi.org/10.1182/blood-2002-10-3175
- Heintel D, Kroemer E, Kienle D, Schwarzinger I, Gleiss A, Schwarzmeier J, Marculescu R, Le T, Mannhalter C, Gaiger A, et al. High expression of activation-induced cytidine deaminase (AID) mRNA is associated with unmutated IGVH gene status and unfavourable cytogenetic aberrations in patients with chronic lymphocytic leukaemia. Leukemia 2004; 18, 756-62; PMID:14961036; http://dx.doi.org/10.1038/sj.leu.2403294
- Abstracts from the XVI International Workshop on Chronic Lymphocytic Leukemia 2015 6 – 9 September 2015 Sydney, Australia. Leuk Lymphoma, 2015; 1-327; PMID:26332288; http://dx.doi.org/10.3109/10428194.2015.1080893