
ABSTRACT
Mitochondrial metabolism is essential to fulfill the large demand for macromolecule biosynthesis in cancer. We recently identified low-level InsP3R-mediated Ca2+ transfer to mitochondria as an unexpected requirement for mitochondrial function. Here we reveal that its absence specifically targets cancer cells and causes necrosis at daughter cell separation during ongoing proliferation.
Otto Warburg suggested that cancer originates from irreversible injury to mitochondria followed by a compensatory increase in glycolysis. Nevertheless, increasing evidence indicates that mitochondrial metabolism is an essential component of most cancer cells and serves as a source of metabolic intermediates for lipid, nucleic acid, and protein synthesis.Citation1 Ca2+ release from the endoplasmic reticulum (ER) through the inositol 1,4,5-trisphosphate receptors (InsP3Rs) regulates numerous cellular functions including gene transcription, cell proliferation, secretion, and motility.Citation2 Recently, we established that low-level constitutive InsP3R-mediated Ca2+ transfer to the mitochondria is essential for maintaining basal levels of oxidative phosphorylation (OXPHOS) and ATP production () in a wide variety of cell types.Citation3 Agonist-induced InsP3R Ca2+ signals enhance mitochondrial function primarily by stimulating the tricarboxylic acid (TCA) cycle dehydrogenases, as well as respiratory chain components, to promote OXPHOS and ATP production.Citation4 In the absence of InsP3R-mediated Ca2+ transfer to mitochondria, cells enter a bioenergetic crisis characterized by a drop in ATP levels, AMPK activation, and induction of mTOR-independent autophagy as an essential survival mechanism.Citation3 () The Ca2+ released by InsP3Rs is taken up by mitochondria via the Ca2+ uniporter (MCU), an inner membrane Ca2+ channel whose molecular identity has recently been revealed.Citation5 The MCU channel is a complex (MCUC) composed of several proteins that regulate its activity, including MCUb, EMRE, MCUR1, MICU1, and MICU2.Citation5 We found that inhibition of mitochondrial Ca2+ uptake by knockdown of MCU or MCUR1 recapitulates the bioenergetic features observed after InsP3R inhibition, namely a decrease in ATP levels, AMPK activation, and induction of prosurvival autophagy (),Citation6,7 thus confirming the essential role of Ca2+ communication between the ER and the mitochondria for cellular homeostasis.
Figure 1. (A) Inhibition of Ca2+ transfer to mitochondria induces cell cycle arrest in normal, but not cancer, cells. Constitutive low levels of InsP3R-mediated Ca2+ transfer to mitochondria maintains the function of the Ca2+-dependent dehydrogenases (PDH, IDH, and KGDH) thus maintaining levels of ATP, NADH, and metabolic intermediates (building blocks) that enable normal and cancer cells to successfully progress through the cell cycle. (B) Suppression of Ca2+ transfer to the mitochondria by inhibition of either InsP3R or MCU decreases TCA cycle activity with concomitant reductions in the levels of NADH, ATP and metabolic building blocks, which activate AMPK and autophagy. Under this metabolic stress normal cells do not engage in the cell cycle whereas cancer cells enter it normally, resulting in mitotic catastrophe and cell death.
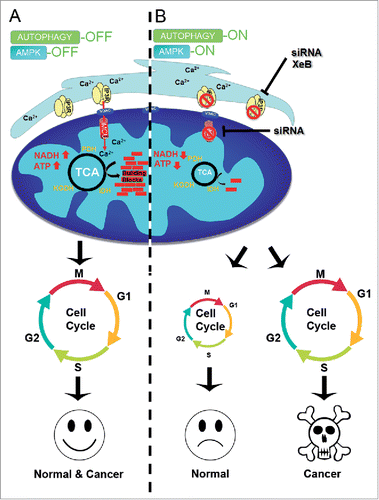
Although increased InsP3R expression and/or activity have been associated with cancer cell proliferation and growth, how InsP3Rs regulate these processes was unknown. Using breast and prostate tumorigenic cell lines and transformed primary human fibroblasts, we discovered that cancer cells also require low-level constitutive InsP3R-mediated Ca2+ transfer to mitochondria to sustain mitochondrial function.Citation8 Genetic or pharmacological inhibition of either InsP3R or MCUC in these cells decreases TCA cycle activity, with a concomitant reduction in the levels of ATP and metabolic intermediates, and activation of AMPK and autophagy. Interestingly, whereas non-cancerous cells are able to survive under these conditions through autophagy, cancer cells die by necrosis at the stage of daughter cell separation during proliferation.Citation8 All the metabolic effects observed due to interruption of ER-to-mitochondria calcium transfer in both normal and cancer cells, as well as the death of cancer cells, were reversed by exogenous addition of the mitochondrial substrates pyruvate or α-ketoglutarate. This protection was unrelated to antioxidant properties of the substrates since NAC was unable to prevent either the metabolic effects or cell death.Citation8 Although AMPK activation in the cancer cells indicates lower ATP levels after inhibition of InsP3R-mediated Ca2+ transfer to mitochondria, much evidence suggests that glycolysis is able to provide enough ATP for cancer cell function.Citation9 Thus, we speculated that in addition to the effects of this Ca2+ transfer on OXPHOS, other mitochondrial functions contribute to the apparent addiction of cancer cells to Ca2+. Considering that at least 3 mitochondrial pathways are involved in pyrimidine and purine nucleotide synthesis and that the rate-limiting step in the de novo synthesis of pyrimidines occurs in the mitochondria,Citation10 we speculated that a lack of metabolic intermediates, in particular nucleotides, is responsible for the observed death of cancer cells. Indeed, nucleoside complementation prevented cancer cell death. Normal cells facing the metabolic stress induced by inhibiting InsP3R-mediated Ca2+ transfer to mitochondria reduced their proliferation by slowing cell cycle progression at the G1/S checkpoint, whereas cancer cells continued to enter the cell cycle regardless of their impaired bioenergetic and metabolic state, leading to death by necrosis at the last step of mitosis (). Treatment of tumors generated by subcutaneous injection of murine melanoma derived B16F10 cells into nude mice with the InsP3R inhibitor Xestospongin B caused marked inhibition of tumor growth associated with massive necrotic cell death without changes in the levels of Ki67, a marker of proliferation, confirming that the cancer cells failed to slow cell growth in the face of their bioenergetic crisis because of a failure to either “monitor” their metabolic status or to engage cell cycle checkpoints, or both. Cell proliferation requires new protein, lipid, and nucleic acid synthesis, and although the interactions between metabolism and the cell cycle are becoming increasingly revealed, our understanding of the molecular wiring that links these processes remains incomplete. Our recent work suggests that inhibition of ER-to-mitochondria Ca2+ transfer uncouples these 2 processes in cancer cells, turning their high proliferative rate, a normally advantageous feature, into a mechanism that promotes their death.
In summary, our recent study suggests that cancer cells are addicted to InsP3R-mediated Ca2+ transfer to mitochondria, and therefore implicates possible therapeutic targets.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grants P30NS047243 (A.L.) and R37GM56328 (J.K.F.), FONDECYT no. 1160332 (C.C.) and FONDAP no. 15150012 (C.C.).
References
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11:325-37; PMID:21508971; http://dx.doi.org/10.1038/nrc-3038
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 2007; 87:593-658; PMID:17429043; http://dx.doi.org/10.1152/physrev.00035.2006
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270-83; PMID:20655468; http://dx.doi.org/10.1016/j.cell.2010.06.007
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012; 51:2959-73; PMID:22443365; http://dx.doi.org/10.1021/bi2018909
- De Stefani D, Patron M, Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 2015; 1853:2006-11; PMID:25896525; http://dx.doi.org/10.1016/j.bbamcr.2015.04.008
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 2012; 14:1336-43; PMID:23178883; http://dx.doi.org/10.1038/ncb2622
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012; 151:630-44; PMID:23101630; http://dx.doi.org/10.1016/j.cell.2012.10.011
- Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep 2016; 14:2313-24; PMID:26947070; http://dx.doi.org/10.1016/j.celrep.2016.02.030
- Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 2008; 18:54-61; PMID:18387799; http://dx.doi.org/10.1016/j.gde.2008.02.003
- Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab 2015; 3:1; PMID:25621173; http://dx.doi.org/10.1186/s40170-015-0128-2