
ABSTRACT
DNA double-strand breaks (DSBs) could be deleterious and lead to age-related diseases, such as cancer. Recent evidence, however, associates DSBs with vital cellular processes. As discussed here, genome-wide mapping of DSBs revealed an unforeseen coupling mechanism between transcription and DNA repair at super-enhancers, as means of hypertranscription of oncogenic drivers.
KEYWORDS:
Despite their reputation as being the most harmful type of DNA damage, more recent studies shed light on the functional roles of DNA double-strand breaks (DSBs) in transcription, replication and genome organization.Citation1–Citation3 Programmed breaks in the DNA support the release of inevitable tension that builds throughout these physiological processes during DNA uncoiling.Citation4
Super-enhancers are large regulatory DNA regions that are responsible for the recruitment and activation of transcription machinery in the cell.Citation5 Oncogenic super-enhancers are defined as those that regulate the transcription of oncogenes and cancer-dependent genes. In cancer, these regions tend to be more active, resulting in higher expression of oncogenes that in turn will promote tumor initiation and progression.Citation6 Areas that are heavily transcribed in the genome are generally less dense, meaning their chromatin is more accessible and exposed. These regions are hence more subject to chromosomal rearrangements leading to genome instability.Citation7 In order to maintain its integrity, transcriptionally active chromatin must be consistently repaired.
In our recent paper,Citation8 published in Cell Reports, the main goal was to map naturally occurring DSBs in their native context, without added induction of external treatments, and later to characterize their role in various cells. The method which was best able to address this goal was BLISS (Breaks labeling in situ and sequencing).Citation9 This method is sensitive enough to identify the locations of DSBs across the genome, as well as to quantify them, allowing us to map the natural “breakome” in the desired cells.
Analysis of the DSBs in different cells revealed the specific breakome pattern of each cell type and highlighted the differences between the cell lines.Citation8 Further analysis categorized which areas of the genome displayed higher break density relative to the entire genome in the different cell lines. It was shown that the highest clustering of breaks occurs at regulatory elements such as insulators, enhancers and promoters. Looking deeper into the category of enhancers in MCF7 breast cancer cells, we found that there is a high density of DSBs at super-enhancers.Citation8
The fact that these breaks were shown to be brought on by transcription led to the idea that there might be a connection between transcription factors and the DSBs. In order to answer that, the genome around the breaks was searched for relevant motifs.Citation8 The TEAD (TEA domain transcription factors) motif, which binds TEAD family transcription factors and is involved in the Hippo pathway, was shown to be enriched around the DSB sites mapped by BLISS. Furthermore, an association between TEAD and AP-1 transcription factors was shown in proximity to MCF7 oncogenic enhancers.Citation8 In order to understand the role of TEAD in genome fragility, chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) for TEAD4 was compared to the MCF7 BLISS-derived breakome. Moreover, The MCF7 breakome was compared to the DSB pattern after depletion of TEAD4. This pinpointed the major correlation between the two to be around strong enhancers only after depletion, suggesting that TEAD4 acts as a protector at these sites and promoting more transcriptional activity, therefore allowing oncogenesis.Citation8
This observation led to the hypothesis that TEAD4 might be partnering with DNA repair factors in order to protect the enhancers. Since RAD51-dependant repair has been previously associated with active transcriptional genes,Citation10 RAD51 was selected to be examined. As with the BLISS, ChIP-seq data showed an enrichment of RAD51 binding sites around regulatory elements.Citation8 These regions also exhibited higher levels of DSBs following RAD51 depletion. Furthermore, the areas that contained overlapping binding sites for TEAD4 and RAD51 were mainly concentrated at strong enhancers, and additional overlap with AP-1 occurred at the sites of super-enhancers which promote the expression of oncogenes in MCF7. Taken together, the observed overlap implies a mechanism which allows ‘on site’ RAD51-dependant repair at areas that are heavily transcribed in order to support them. It was further determined that the areas most affected by RAD51 depletion were the RAD51/TEAD4 common sites. Moreover, inhibition of RAD51 activity revealed reduced transcription of enhancer RNA (eRNA) at these sites, as determined by RNA sequencing.Citation8
What causes the onset of the DSBs? Transcription-related programmed DSBs are mediated via topoisomerases (TOPs).Citation4 ChIP-seq of TOP1, but not TOP2b, revealed a high overlap with RAD51 binding sites at strong enhancers. Further, genomic regions enriched with TOP1 binding display more BLISS signal and inhibition of TOP1 activity showed an increase of DSBs at these sites, suggesting that TOP1 is involved in RAD51-associated DSBs.Citation8
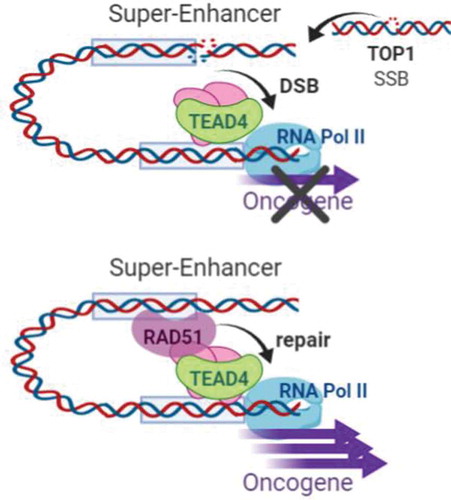
Programmed DNA damage has been documented in actively transcribed regions facilitating, for example, B-cell receptor diversification and chromosome folding.Citation7 Faulty repair of these regions could lead to catastrophic consequences posing a threat to genomic stability. The work described here shows a novel mechanism through which cancer cells “hijack” the DNA damage response (DDR) in order to allow heavy transcription, thus protecting the integrity of their genome and supporting tumor progression (). These findings imply that inhibition of RAD51, and likely other DDR factors, could heavily alter transcription leading to a wide range of catastrophic events.
Figure 1. Hypertranscription of oncogenes regulated by super-enhancers is coupled by a RAD51-dependent repair mechanism. Mapping of the breakome reveals enrichment of DSBs in super-enhancers. These breaks are likely caused by TOP1 leading to single strand breaks (SSBs) that develop into DSBs. Repair of these breaks is mediated by RAD51 and results in high transcription of oncogenic drivers.
The outcomes of this study also propose a therapeutic strategy involving the inhibition of both TEAD4 and RAD51. Given that RAD51 depletion resulted in a larger effect on the cancerous MCF7 cells in comparison to the effect on the pre-malignant MCF10A cells,Citation8 entails additional exploration of this route. Future work shall uncover the relevance of this approach in using other DDR drugs such as PARP inhibitors. Moreover, further investigation is needed into the similarity of this mechanism in other malignant cell types, hopefully providing a broader therapeutic opportunity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Madabhushi R, Gao F, Pfenning A, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao P-C, et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161:1–2. doi:10.1016/j.cell.2015.05.032.
- Bunch H, Lawney BP, Lin Y-F, Asaithamby A, Murshid A, Wang YE, Chen BPC, Calderwood SK. Transcriptional elongation requires DNA break-induced signalling. Nat Commun. 2015;6:10191. doi:10.1038/ncomms10191.
- Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon K-R, Pekowska A, Zhang H, Rao SSP, et al. Genome organization drives chromosome fragility. Cell. 2017;170:507–521 e518. doi:10.1016/j.cell.2017.06.034.
- Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9:327–337. doi:10.1038/nrc2608.
- Whyte WA, Orlando D, Hnisz D, Abraham B, Lin C, Kagey M, Rahl P, Lee T, Young R. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi:10.1016/j.cell.2013.03.035.
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi:10.1126/science.1259037.
- Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–656. doi:10.1016/j.cell.2017.01.002.
- Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of oncogenic super-enhancers is coupled with DNA repair by RAD51. Cell Rep. 2019;29:560–572. doi:10.1016/j.celrep.2019.09.001.
- Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017;8:15058. doi:10.1038/ncomms15058.
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. doi:10.1038/nsmb.2796.