
ABSTRACT
When the orthologue of tumor suppressor protein p53 (TP53), cep-1, is inactivated in Caenorhabditis elegans, the nematodes manifest an autophagy-dependent increase in lifespan. A recent paper by Yang et al. demonstrates that accelerated aging phenotype of autophagy-deficient mice can be reduced by the knockout (KO) of Trp53. These findings point to a complex bidirectional crosstalk between autophagy and TP53 that has vast implications for the aging process.
Tumor suppressor protein p53 (TP53) is a widely studied protein with multiple functions. This tumor suppressor is mutated or functionally inactivated in the majority of human cancers. TP53 is also a stress-inducible transcription factor for genes involved in metabolism, autophagy, senescence, apoptosis, cell cycle arrest, and DNA repair. TP53 activates a series of distinct pathways that define cell fate depending on the context and the nature of the upstream stressors.Citation1 TP53 can temporarily stop the cell cycle to allow repair mechanisms to intervene, ensuring the survival of the cell. Conversely, if the damage is irreversible, cells undergo apoptosis or enter a metabolic dormant state known as senescence, where their cell cycle remains blocked. Thus, the outcome of DNA damage repair can lead to survival, death, tumorigenic transformation (when cells elude TP53 checkpoints), or to the blockade of the cell cycle when damage is persistent. In addition, TP53 has been postulated to have a role in regulating autophagy, a survival mechanism responsible for the removal of damaged and potentially dangerous cellular compartments.
During aging, a drop in autophagy and the accumulation of senescent cells prove to be detrimental to the organism, as stem cells enter senescence causing a progressive defect of the regenerative machinery.Citation2 Moreover, the neutralization of TP53 can alleviate signs of aging, prolonging the lifespan of nematodes.Citation3 Given their role in cellular homeostasis and aging, an intimate interconnection between TP53 and autophagy has been postulated.Citation3
A recent paper by Yang et al. in Genes & DevelopmentCitation4 adds important insights into the relationship between autophagy and TP53 in the context of aging. Several years ago, Eileen White’s group reported that the inducible full-body knockout (KO) of the essential autophagy gene Atg7 causes a spectacular acceleration of aging in most if not all tissues, as well as premature death of mice due to neurodegeneration.Citation5 Now, the same group has reported that the simultaneous deletion of both Atg7 and Trp53 prolongs lifespan as compared to the Atg7 KO mice, by delaying neurodegeneration in one mouse out of three.Citation4 Trp53 KO mice may live up to 6 months, but prematurely die from thymic lymphoma. In Atg7/Trp53 double KO mice, such tumors rarely develop, suggesting that Atg7 plays a role in promoting thymic lymphomas. Moreover, Atg7/Trp53 double KO mice were more resistant to fasting compared to Atg7 KO mice, suggesting that the TP53-inducible genes contribute to the lethal outcome of fasting in Atg7-deficient mice. Indeed, fasting of Atg7-deficient animals results in the upregulation of TP53 pro-apoptotic target genes (such as Cdkn1a, Bax, Bbc3), suggesting a mechanism for this connectionCitation4 ().
Figure 1. Relationship between aging, autophagy, and TP53. a. Autophagy inhibition causes enhanced transactivation of tumor suppressor protein p53 (TP53) target genes, resulting in accelerated aging of mice. b. TP53 inactivation results in enhanced autophagy (due to the absence of cytoplasmic TP53), increasing longevity in nematodes. c. Hypothetical experimental design to distinguish the roles of cytoplasmic and nuclear TP53 in the aging process. It would be necessary to generate TP53 mutants that are selectively active in the cytoplasm or in the nucleus, to generate genetically modified mice expressing such mutants and to characterize them for age-associated traits.
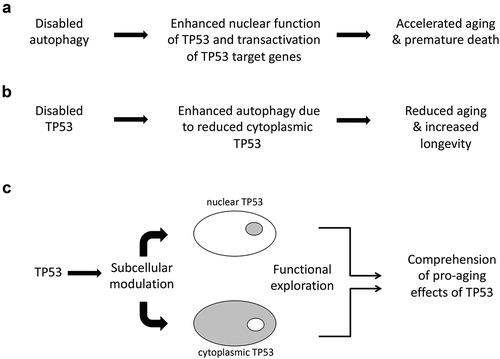
Previous work by Tasdemir et al.Citation6 led to the discover that a non-nuclear, cytoplasmic pool of TP53 regulates autophagy. In the context of cancer, an intertwined regulation between autophagy and cytoplasmic TP53 exists. Most notably, the depletion of cytoplasmic TP53 facilitates autophagy induction, while stabilization of cytoplasmic TP53 can restrain autophagy in Trp53 KO mice and prevent ER stressor-induced activation of autophagy in cancer cells. Moreover, by as yet elusive mechanisms, autophagy favors nuclear export of TP53, ultimately favoring its proteasomal degradation.Citation6 More importantly, in Caenorhabditis elegans, KO of the TP53 orthologue cep-1, results in a Beclin 1-dependent increase in both autophagy and longevity,Citation3 indicating that inhibition of autophagy by TP53 accelerates aging ().
Inhibition of autophagy by RNA interference targeting Atg5 induces markers of senescence including high expression of TP53,Citation7 in the kidney, heart, liver, and muscle from mice, confirming the essential role of autophagy in organismal homeostasis and the avoidance of excessive TP53 accumulation. Conversely, recent findings reveal that Sestrins (which can be transactivated by TP53) upregulate autophagy as a protective mechanism for muscles undergoing atrophy and aging.Citation8
Baar et al.Citation9 identified FOXO4 as a TP53 interactor that contributed to the long-term survival of senescent cells by inhibiting the TP53-mediated transactivation of pro-apoptotic genes and rather favoring cell cycle arrest. With the aim to develop specific senolytic agents, they designed peptides that disrupt the interaction between TP53 and FOXO4. In pre-aged mice, this maneuver caused the exclusion of TP53 from the nucleus, and its subsequent accumulation in the cytosol, priming senescent cells to Caspase-3/7 dependent apoptosis,Citation9 pointing to the possibility that cytoplasmic TP53 would acquire the capability to promote senolysis, the selective removal of senescent cells for the avoidance of aging.
Altogether, these findings plead in favor of a dual role of TP53 in the regulation of senescence, depending on the model and its subcellular localization. It will be important to understand whether the inhibition of a prosurvival mechanism such as autophagy by cytosolic TP53 would interfere with (or rather favor) the age-associated accumulation of senescent cells. To clarify the complexity of the crosstalk between TP53 and autophagy, it would be useful to create inducible knock-in mouse models in which TP53 can be switched to exclusively nuclear versus cytoplasmic locations ().
Disclosure of Potential Conflicts of Interest
G.K. has been holding research contracts with Bayer Healthcare, Genentech, GlaxoSmithKline, Institut Mérieux, Kaleido, Lytix Pharma, Nucana, Oncolinx, PharmaMar, Samsara, Sotio, and Vasculox. G.K. is on the Board of Directors of the Bristol-Myers Squibb Foundation France. G.K. is a scientific co-founder of everImmune, Samsara Therapeutics, and Therafast Bio.
Acknowledgments
V.S. acknowledges funding from a FEBS Long-Term Postdoctoral Fellowship and the “Unidad de Excelencia María de Maeztu”, funded by the AEI (CEX2018-000792-M).
G.K. is supported by: the Ligue contre le cancer (Équipe Labellisée); Agence National de la Recherche (ANR) – Projets Blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la Recherche sur le Cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des Universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the EU Horizon 2020 Project Oncobiome; Fondation Carrefour; High-End Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
References
- Labuschagne CF, Zani F, Vousden KH. Control of metabolism by p53 - Cancer and beyond. Biochim Biophys Acta Rev Cancer. 2018;1870(1):1–3. doi:10.1016/j.bbcan.2018.06.001.
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(7584):37–42. doi:10.1038/nature16187.
- Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy. 2008;4(7):870–873. doi:10.4161/auto.6730.
- Yang Y, Karsli-Uzunbas G, Poillet-Perez L, Sawant A, Hu ZS, Zhao Y, Moore D, Hu W, White E. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 2020;34(9–10):688–700. doi:10.1101/gad.335570.119.
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. doi:10.1158/2159-8290.CD-14-0363.
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. doi:10.1038/ncb1730.
- Cassidy LD, Young ARJ, Young CNJ, Soilleux EJ, Fielder E, Weigand BM, Lagnado A, Brais R, Ktistakis NT, Wiggins KA. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat Commun. 2020;11(1):307. doi:10.1038/s41467-019-14187-x.
- Segalés J, Perdiguero E, Serrano AL, Sousa-Victor P, Ortet L, Jardí M, Budanov AV, Garcia-Prat L, Sandri M, Thomson DM. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat Commun. 2020;11(1):189. doi:10.1038/s41467-019-13832-9.
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1):132–147.e16. doi:10.1016/j.cell.2017.02.031.