
ABSTRACT
The phosphatidylinositol 3-kinase (PI3K), which is composed of the p85 regulatory and p110 catalytic subunits, is known to be downstream of the receptor tyrosine kinase (RTK). Our recent findings revealed that p85β increases the protein level of AXL (an RTK) to activate p110, suggesting bidirectional regulation between PI3K and RTK.
KEYWORDS:
The class IA phosphoinositide-3 kinase (PI3K) is considered as a major lipid kinase that phosphorylates PI(4,5)P2 to produce PI(3,4,5)P3.Citation1 While the p110 subunit of the PI3K heterodimer catalyzes the reaction, the p85 regulatory subunit stabilizes and inhibits p110. Earlier studies have focused on mutations of PIK3CA (p110α) or loss of PTEN for PI3K pathway hyperactivation in cancers, possibly because of the high aberration frequencies and their direct enzymatic effects on the phospholipids. Until recently, abnormal signaling caused by aberrations of the p85 regulatory isoforms has been revealed. p85α (PIK3R1) functions as a tumor suppressor in multiple cancer lineages, but p85β (PIK3R2) has been suggested as an oncoprotein. Increased p85β level was previously observed in breast and colon carcinomas, which consequently showed enhanced PI(3,4,5)P3 levels and PI3K pathway activation.Citation2 However, the oncogenic mechanisms of p85β are yet to be characterized.
In our recent study, the oncogenicity of p85β was demonstrated in ovarian cancer.Citation3 Increases in p110 kinase activity and PI(3,4,5)P3 level were consistently observed whereas the protein level of p110 was not altered, suggesting that the promotion of PI3K activity is not due to an enhanced p110 protein stability. Strikingly, through a protein array that targets major cancer pathways, we identified AXL as a mediator that accounts for p110 activation and the oncogenic signaling induced by p85β. AXL is a receptor tyrosine kinase (RTK) of the TAM family, in which the other members are Tyro3 and MERTK. AXL, initially identified as a transforming gene isolated from chronic myeloid leukemia patients, is upregulated and is recognized as a biomarker of poor prognosis in several cancer types including that of the ovary.Citation4 AXL protein level was markedly upregulated in more than 80% of stage III high-grade or metastatic serous ovarian cancer patients.Citation4,Citation5 Functionally, we confirmed that inhibition of AXL substantially blocked the tumorigenic capabilities and p110 activity induced by p85β.
Transcriptional, post-transcriptional, protein degradation pathways have been reported as the regulatory mechanisms of AXL (). Indeed, AXL amplification is uncommon in cancers, for example less than 3% of The Cancer Genome Atlas (TCGA) ovarian cancer cases (n = 585) was AXL-amplified. Our analyses showed positive association between the protein levels of p85β and AXL in the TCGA and an independent ovarian cancer datasets. Regulation of AXL protein stability but not AXL promoter or transcript by p85β was validated in ovarian cancer cell lines, indicating a post-translational regulation. Remarkably, in contrast to p110 which is stabilized by p85 through inhibiting thermal degradation,Citation6 p85β stabilizes AXL protein through inhibiting the autophagy-lysosomal degradation pathway. First, we demonstrated that lysosomal but not proteasomal inhibitors were able to reverse AXL degradation upon p85β depletion. Second, accompanied with AXL degradation was an increased co-localization between AXL protein and key autophagy/lysosome markers. Autophagic flux was also induced upon p85β silencing. Third, TRIM2 (an E3 ligase) and optineurin (an autophagy receptor) were identified to mediate the entry of AXL into the autophagy pathway. Importantly, p85β likely regulates the activities of TRIM2 and optineurin by altering the phosphorylation of these two molecules. It would be interesting to further determine the other proteins that are regulated by p85β through the autophagy pathway. TRIM2 is a new ligase for AXL. Previously identified ligases include c-Cbl which mediates AXL degradation induced by the TAM ligand Gas6 and Cbl-b which targets all the three TAM receptors.Citation7,Citation8 c-Cbl and Cbl-b are not involved in AXL regulation by p85β. This may align with our observations that the regulation by p85β was Gas6-independent and was highly selective to AXL. Protein levels of Tyro3 and MERTK were not altered by p85β. Intriguingly, our data supported that the regulation of AXL by p85β was independent of p110 kinase activity because neither pan-p110 nor p110 isoform-specific inhibitors abolished the regulation. p85β lacks intrinsic kinase activity. How p85β affects the phosphorylation of TRIM2 and optineurin remains unknown. We have previously described the formation of p110-free p85α homodimer that binds PTEN and inhibits PTEN degradation.Citation9 In addition, p85α may possess GTPase-activating protein (GAP) activity toward Rab GTPases which in turn control trafficking and recycling of RTKs such as platelet-derived growth factor receptor.Citation10 In light of these findings, whether p110-independent “free” p85β molecules exist and whether p85β regulates AXL through GAP warrant future investigations.
Figure 1. Molecular mechanisms of AXL regulation. Several transcription factors that regulate AXL promoter have been identified. AXL mRNA can also be regulated by tumor suppressive miRNA such as miR-34a and miR-199a/b. At post-translation level, AXL protein is regulated through proteolytic shedding of the extracellular domain or through protein degradation mediated by E3 ligases. In our recent study, p85β inhibits autophagic degradation of AXL by modulating the phosphorylation of E3 ligase TRIM2 and autophagic receptor optineurin.
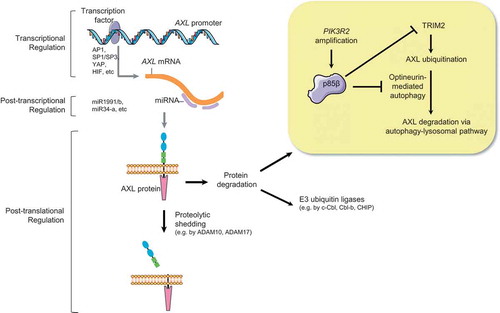
Our results collectively indicate that p85β increases AXL protein level to activate p110. One of the primary roles of p85 is to inhibit p110 activity in quiescent state. It has been shown that cancer cells derive strategies to overcome the inhibition, for examples, through point mutations of PIK3CA or PIK3R1 that relieve the inhibitory interaction. Point mutation of PIK3R2 is rare in cancers, therefore PIK3R2 amplification could be an alternative strategy leading to p110 activation. We investigated the oncogenic role of p85β in ovarian cancer because PIK3R2 amplification is commonly seen in high-grade serous ovarian cancer (amplification frequency: 50% of TCGA samples) and high PIK3R2 levels correlate with worse patient survival. Our drug response data revealed that p85β-expressing xenograft tumors were more vulnerable to inhibition of AXL than control tumors. Therefore, PIK3R2 amplification or p85β level could be a potential biomarker of responsiveness to AXL-targeting therapy in ovarian cancer. In fact, PIK3R2 amplification is also frequent in glioblastoma (40%), breast carcinoma (20%) and endometrial carcinoma (17%) in the TCGA cohorts. The functional impacts and therapeutic implications of PIK3R2 amplification in these cancer types remain to be explored.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1–3. doi:10.1126/science.296.5573.1655.
- Cortés I, Sánchez-Ruíz J, Zuluaga S, Calvanese V, Marques M, Hernandez C, Rivera T, Kremer L, Gonzalez-Garcia A, Carrera AC, et al. p85β phosphoinositide 3-kinase subunit regulates tumor progression. Proc Natl Acad Sci USA. 2012;109(28):11318–11323. doi:10.1073/pnas.1118138109.
- Rao L, Mak VC, Zhou Y, Zhang D, Li X, Fung CCY, Sharma R, Gu C, Lu Y, Tipoe GL, et al. p85β regulates autophagic degradation of AXL to activate oncogenic signaling. Nat Commun. 2020;11(1):2291. doi:10.1038/s41467-020-16061-7.
- Lozneanu L, Pinciroli P, Ciobanu DA, Carcangiu ML, Canevari S, Tomassetti A, Căruntu I-D. Computational and immunohistochemical analyses highlight AXL as a potential prognostic marker for ovarian cancer patients. Anticancer Res. 2016;36:4155–4163.
- Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, Wei K, Kuo CJ, Longacre TA, Giaccia AJ, et al. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70(19):7570–7579. doi:10.1158/0008-5472.CAN-10-1267.
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3ʹ-kinase: stabilization and inhibition of the p110α catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18(3):1379–1387. doi:10.1128/MCB.18.3.1379.
- Valverde P. Effects of Gas6 and hydrogen peroxide in AXL ubiquitination and downregulation. Biochem Biophys Res Commun. 2005;333(1):180–185. doi:10.1016/j.bbrc.2005.05.086.
- Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508–512. doi:10.1038/nature12998.
- Cheung LW, Walkiewicz KW, Besong TM, Guo H, Hawke DH, Arold ST, Mills GB. Regulation of the PI3K pathway through a p85α monomer–homodimer equilibrium. Elife. 2015;4:e06866. doi:10.7554/eLife.06866.
- Chamberlain MD, Oberg JC, Furber LA, Poland SF, Hawrysh AD, Knafelc SM, McBride HM, Anderson DH. Deregulation of Rab5 and Rab4 proteins in p85R274A-expressing cells alters PDGFR trafficking. Cell Signal. 2010;22(10):1562–1575. doi:10.1016/j.cellsig.2010.05.025.