
ABSTRACT
A multitude of ultrafast photoinduced reactions in organic semiconductors are governed by the close interplay between nuclear and electronic degrees of freedom. From biological light-harvesting and photoprotection to organic solar cells, the critical electronic dynamics are often precisely synchronized with and driven by nuclear motions, in a breakdown of the Born-Oppenheimer approximation. Ultrafast time-domain Raman methods exploit impulsive excitation to generate nuclear wavepackets and track their coherent evolution through these reaction pathways in real time. This tool of vibrational coherence has recently been applied to study singlet fission, a carrier multiplication process with the potential to boost solar cell efficiencies which has been under intense mechanistic investigation for the past decade. In this review, we present the essential features of the spectroscopic techniques and discuss how they have been used to elaborate a new perspective on the singlet fission mechanism. It is now established that ultrafast triplet-pair formation is driven by vibronic coupling, whether fission is exothermic or endothermic, and thus that full understanding of singlet fission requires explicit consideration of nuclear dynamics. Despite broad qualitative agreement between different vibrational coherence methods, differences in the detailed observations and interpretation raise important questions and pose new challenges for future research.
Graphical abstract

1. Introduction
Photoinduced processes in organic materials are often explained by the simple transfer of an electron from reactant to product states under the adiabatic Born-Oppenheimer approximation, assuming essentially static nuclei during the fast movement of an electron. In this regime, the primary role of molecular vibrations is in energy dissipation, functioning as a molecular ‘brake’. For many physically relevant systems, the gap between electronic states approaches vibrational energies and this approximation begins to break down. The resulting non-adiabatic regime yields coherent evolution of nuclear and electronic wavefunctions, and the intrinsic ‘floppiness’ of molecules becomes an asset that can drive fast and efficient dynamics. Small nuclear coordinate changes can then guide the outcomes of electronic processes, as seen in cis-trans photoisomerization of retinal in vision [Citation1], photochemical bond dissociation [Citation2] and the protection mechanisms of DNA under UV light exposure [Citation3]. Similar coherent coupling of vibrational and electronic states is recognized as a driver in numerous photoinduced processes in which the excitation energy is maintained and transformed between different excited electronic states [Citation4], for instance biological light harvesting [Citation5], electron transfer in donor-acceptor molecules [Citation6] and charge generation in organic photovoltaics [Citation7].
Such vibronic coupling between electronic and nuclear degrees of freedom and the complex potential energy surfaces (PESs) that results are indeed long established to play a crucial role in the electronic processes in molecular materials [Citation8]. These ideas have even been exploited as a gateway to actively control or redirect electronic processes through the manipulation of particular vibrational states [Citation9–12]. Nevertheless, the concept of vibronic coupling rarely enters explicitly into the pictures drawn of photophysics in complex systems. Instead, complex electronic dynamics are frequently described in terms of simple ‘stick’ diagrams and the implicit simplifying assumption of static nuclei. The cases highlighted above, however, demonstrate that the detailed mechanism of ultrafast processes must explicitly consider the nuclear motions as well. This demands the use of more sophisticated spectroscopic methods, sensitive to both electronic and nuclear dynamics. Where this challenge is met, entirely new levels of understanding can be achieved.
As a case in point, in this review we will address how these ideas have influenced the field of singlet fission (SF). SF is a carrier multiplication phenomenon unique to organic semiconductors in which a photoexcited singlet state S1 can separate into a pair of distinct triplets. The fundamental requirements of the process are energy conservation – requiring that E(S1)~2E(T1), allowing for some contributions from thermal energy – and spin conservation. The former can be achieved through molecular design, while to satisfy the latter the triplets must be initially produced in a spin-entangled overall singlet configuration. Originally treated as a theoretical abstraction to mediate singlet-triplet interconversion [Citation13,Citation14], the entangled triplet pair 1TT is now recognized as a distinct electronic state with unique photophysical properties [Citation15–17]. Indeed, a wide range of triplet-pair states of different spin or boundedness have been detected and invoked [Citation18–21], leading to the general kinetic scheme in (). The principal motivation to study SF in recent years has been to harness its potential to overcome the Shockley-Quiesser limit on the efficiency of single-junction solar cells[Citation22]. This endeavour calls for a more detailed understanding of the molecular SF mechanism, which has inspired the application of a wide range of spectroscopic and computational tools to the relatively small family of known SF materials. We point the interested reader to other detailed reviews on the historic development of the field [Citation14,Citation23], SF device applications [Citation24,Citation25], the properties of triplet-pair states [Citation16,Citation26–28], theoretical modelling [Citation29] and molecular design principles [Citation30,Citation31]. We focus here on mechanistic investigations and how they have been impacted by new understandings of vibronic coupling.
Figure 1. Standard schematic of the multi-step SF process. The focus of this review is the initial S1→1TT conversion, where vibronic coupling mechanisms have been implicated
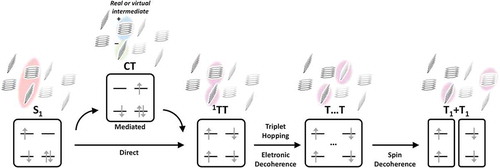
For most of the past decade, the principal debate in the SF field has been the nature of the initial step in SF, now defined as S1→1TT conversion, which can occur coherently or incoherently. In early years, the focus was on the low-lying electronic states in canonical systems like pentacene and how they relate to diabatic S1, 1TT and charge-transfer (CT) excitations. The strength of the electronic coupling between these configurations was considered to determine the rate and efficiency of triplet formation through one of several mechanisms with equations typically for incoherent SF describing stochastic population hopping from S1 to 1TT. In ‘mediated’ SF, S1 couples most strongly to CT states; these provide effective coupling to 1TT, serving as a bridge between manifolds whether directly populated [Citation14,Citation32] or only virtually involved [Citation33–36]. In the alternative ‘direct’ model, the two-electron coupling between S1 and 1TT is sufficient to drive population transfer with no intermediates [Citation37–39].
Coherent SF can be divided into two perspectives. First, under extreme S1-1TT or S1-CT and CT-1TT coupling, we obtain the ‘quantum coherent’ mechanism. Here, S1 and 1TT are linked from the instant of photoexcitation by pure electronic coherence, giving rise to the generation of a quantum superposition of S1 and 1TT (indirectly coupled via CT) and thereby enabling ultrafast 1TT formation[Citation40]. In subsequent work, this model was refined to include indirect coupling bewith few exceptions [Citation37,Citation38,Citation41], such models focused on fixed nuclear geometries with little explicit role for vibrations. The same ideas of static, diabatic S1 and 1TT states permeated most spectroscopic work. Time-resolved techniques, whether photoluminescence, transient absorption or frequency-domain Raman, identified the electronic signatures of S1 and T1 and the kinetics of interconversion. The resulting timescales could often be described by multiple competing theories, and thus the principal tools for tracking the SF reaction provided little insight into the core questions about the nature of the relevant couplings. The chief exception, the breakthrough report of instantaneous coherent 1TT formation using photoemission spectroscopy, which implied very strong electronic coupling [Citation42,Citation43], subsequently proved controversial [Citation44,Citation45]. A broad study of structure-dependent SF rates in polyacene materials formulated an adiabatic mechanism of continuously evolving wavefunction character from S1 to 1TT along an arbitrary reaction coordinate, but no explicit link was made to nuclear dynamics[Citation44].
This perspective changed with a pair of breakthrough studies applying time-domain Raman spectroscopy to ultrafast (<100 fs) SF in TIPS-pentacene, pentacene and DT-pentacene, probing the fate of wavepackets of vibrational coherence (VC) [Citation46,Citation47]. These works directly monitored real-time coherent vibrational motions synchronized with the electronic dynamics of SF and identified VC in the product 1TT state, demonstrating an intimate link between these nuclear motions and the ultrafast reaction. This has been called the ‘vibronic coupling’ or ‘vibrationally coherent’ mechanism, which corresponds to the case that S1 and 1TT are nonadiabatically coupled through vibrational coordinates or form vibronically mixed states when their energy levels are resonant owing to excited vibrational quanta. This is the framework in which the earlier quantum coherent results are now understood as well. A full understanding of ultrafast SF thus requires explicit consideration of nuclear dynamics, and numerous studies have exploited the real-time detection of coherent nuclear motions on excited-state PESs to extend this paradigm to other SF systems. The role of vibronic coupling between S1 and 1TT states to trigger coherent SF is now firmly established [Citation41,Citation48–53]. In this review, we address recent developments in time-domain Raman techniques and their role in advancing vibronic coupling mechanisms for ultrafast 1TT formation. Despite the rapid early progress using these methods, we highlight a number of critical gaps in the field that call for further research.
2. Basics of time-domain Raman spectroscopy
We first provide a brief introduction to the time-domain Raman spectroscopy methods that have been applied to the study of SF. Our aim here is to bring out the core concepts on an introductory level suitable to interpret the SF results. The interested reader can find fuller technical details in a recent review by Buckup and Léonard [Citation54] and the works cited below.
Time-domain Raman spectroscopy – first introduced as impulsive stimulated Raman scattering (ISRS) pioneered by Nelson and co-workers in 1985 – [Citation55] has enabled the direct measurement of coherent molecular vibrations/phonons through the time-domain signals of vibrational wavepackets, that is, VC, corresponding to the coherent superposition of multiple vibrational eigenstates [Citation56,Citation57]. The relative phase between these eigenstates results in oscillations of the sublevel populations, at frequencies determined by the vibrational spacing. The essential prerequisite to generating VC is an excitation laser pulse which is temporally short relative to the vibrational periods of interest ()).
Figure 2. (a) Typical Raman spectrum of organic molecules, indicating the corresponding timescale which informs the necessary pulse duration to generate VC. (b) VC-induced oscillations manifested in transient absorption spectra and kinetics. (c) Generation of ground-state VC via the ISRS mechanism under off-resonant impulsive excitation. This mechanism remains active under resonant conditions. (d) Generation of excited-state VC through resonant impulsive excitation. (e) Typical absorption spectra of an organic molecule showing structured vibronic bands and simulated Gaussian impulsive pulses with <10 fs transform limit. The top and bottom spectra correspond to panels a and b. In the intermediate partially resonant condition, excited-state VC can be observed but it will be predominantly low-frequency due to poor spectral overlap. Adapted from [63], copyright 2015 American Chemical Society
![Figure 2. (a) Typical Raman spectrum of organic molecules, indicating the corresponding timescale which informs the necessary pulse duration to generate VC. (b) VC-induced oscillations manifested in transient absorption spectra and kinetics. (c) Generation of ground-state VC via the ISRS mechanism under off-resonant impulsive excitation. This mechanism remains active under resonant conditions. (d) Generation of excited-state VC through resonant impulsive excitation. (e) Typical absorption spectra of an organic molecule showing structured vibronic bands and simulated Gaussian impulsive pulses with <10 fs transform limit. The top and bottom spectra correspond to panels a and b. In the intermediate partially resonant condition, excited-state VC can be observed but it will be predominantly low-frequency due to poor spectral overlap. Adapted from [63], copyright 2015 American Chemical Society](/cms/asset/5d59ddb6-4d00-4ef5-aea8-0ebb027fc007/tapx_a_1918022_f0002_oc.jpg)
According to the time-energy uncertainty principle, this is equivalent to requiring an energetically broad excitation, suitable to span multiple vibrational levels. The pulse width thus directly determines the observable frequency window.
The simplest case of time-domain Raman spectroscopy is a pump-probe experiment, typically called impulsive vibrational spectroscopy (IVS) or broadband transient absorption (BBTA) [Citation58–63]. Here, the impulsive pump pulse generates VC, which manifests as oscillatory modulations to the standard transient signatures of the ground and excited electronic states, namely ground-state bleaching (GSB), stimulated emission (SE), and excited-state absorption (ESA)/photo-induced absorption. Such spectral effects of VC can be monitored through the transmission of a broadband probe pulse ()). Fourier transformation is typically used to convert time-domain VC signals into frequency-domain Raman spectra. Direct analysis of time-domain information by itself, such as amplitude, phase, or dephasing time, can provide further valuable information.
The general condition of ultrafast/broadband excitation can generate VC both in the ground and the excited electronic states. This can pose a crucial challenge, as it is specifically excited-state VC which presents the major interest in ultrafast photophysics and photochemistry. The intensity of excited-state relative to ground-state VC strongly depends on the resonance condition, the spectral overlap between the pump pulse and the sample absorption spectrum. When spectral overlap is poor or absent, corresponding to the pre- or off-resonant condition, VC is only created on the ground-state PES through the ISRS mechanism () and the top of )). A widespread example of this phenomenon is the appearance of strong solvent VC signatures in solution-phase measurements [Citation64].
At the other extreme, under the fully resonant condition where the pump entirely covers the absorption spectrum, excited-state VC can be efficiently generated () and the bottom of )). Here, to create high-frequency excited-state VC in the typical Raman fingerprint region, corresponding to single- or double-bond vibrations, the pump pulse must have sufficient bandwidth to span higher vibrational sublevels (vibronic bands) of the sample absorption, typically >1500 cm−1 corresponding to a duration of <25 fs ()). The combination of bandwidth and overlap is key – if a suitably broadband pulse only partially overlaps with the ground-state absorption (the middle of )), any resulting high-frequency VC is likely to belong to the ground state. Even in optimal conditions, though, the generation of ground-state VC cannot be avoided, and isolating pure excited-state VC from the entangled oscillation signals that result has long been regarded as one of the chief issues facing time-domain Raman methods[Citation65].
A common tool to identify excited-state VC is through its spectral position. Assuming that the fully resonant condition is satisfied, excited-state VC is most likely to predominate in the ESA or SE region, since these signatures reflect purely excited-state transitions ()). The dominant contribution in the GSB spectral region, on the other hand, should be ground-state VC. Even when the Stokes shift is very small such that GSB and SE signals are significantly overlapped, probe-wavelength-dependent Fourier amplitude, oscillation phase, or damping time analysis can differentiate between the ground- and the excited-state VCs [Citation57,Citation66]. Such spectral analysis must be used with caution, however; ground-state VC signatures, including of solvent molecules with no mechanistic role, can often be recorded across the entire probe detection window [Citation46,Citation64]. Where possible, purely optical means to isolate excited-state VCs are preferred as they are less susceptible to analytical bias. For instance, the chirp of pump pulses can be tuned to selectively enhance the intensity of VCs on either the ground or excited state [Citation67,Citation68]. Alternatively, an additional ‘population control’ pulse can be incorporated within the pump-probe IVS sequence following the broadband impulsive pump and resonant with an SE or ESA transition. This pulse selectively modifies the VC of the targeted electronic state and enables exclusive isolation of pure excited-state signals[Citation69]. Thus, while distinguishing ground- and excited-state VC remains a key challenge in the analysis of time-domain Raman spectroscopy, recent technical and analytical developments have provided experimentalists a range of options to achieve this.
Figure 3. Experimental schemes of time-domain Raman spectroscopic techniques and their representative data. (a) IVS and DFWM. Representative wavelength-resolved impulsive Raman map of β-carotene obtained by IVS reveals vibrational spectra of S1 (bottom) and S2 (top) states. Adapted from [113], copyright 2014 American Physical Society. (b) 2DES. Experimental 2D beating maps of PC645 protein in algae (top) can be theoretically simulated based on whether the VCs are purely vibrational (bottom left) or vibronic (bottom right) in origin. Adapted from [80], copyright 2016 Elsevier Inc. (c) Pump-IVS and Pump-DFWM. Time-resolved Raman spectra of a donor-acceptor-donor-type perylene diimide dye obtained by Pump-IVS reveals continuous evolution of vibrational spectrum during charge separation. Adapted from [85], copyright 2020 Wiley-VCH Verlag GmbH &Co. KGaA
![Figure 3. Experimental schemes of time-domain Raman spectroscopic techniques and their representative data. (a) IVS and DFWM. Representative wavelength-resolved impulsive Raman map of β-carotene obtained by IVS reveals vibrational spectra of S1 (bottom) and S2 (top) states. Adapted from [113], copyright 2014 American Physical Society. (b) 2DES. Experimental 2D beating maps of PC645 protein in algae (top) can be theoretically simulated based on whether the VCs are purely vibrational (bottom left) or vibronic (bottom right) in origin. Adapted from [80], copyright 2016 Elsevier Inc. (c) Pump-IVS and Pump-DFWM. Time-resolved Raman spectra of a donor-acceptor-donor-type perylene diimide dye obtained by Pump-IVS reveals continuous evolution of vibrational spectrum during charge separation. Adapted from [85], copyright 2020 Wiley-VCH Verlag GmbH &Co. KGaA](/cms/asset/25827c17-b139-4132-a8ef-b36fba0f3109/tapx_a_1918022_f0003_oc.jpg)
Comparable time-domain vibrational information can also be achieved with other techniques. Degenerate-four-wave-mixing (DFWM) or transient grating (TG) spectroscopy uses broadband pumping to generate similar VC signals as in the case of IVS, and these are read out as oscillations in the subsequent probe-detected signal [Citation70–73]. The crucial distinctions between IVS and DFWM arise from the latter being performed in a BOXCARS geometry ()). In IVS, the two-field interaction necessary to generate VC is accomplished with the single broadband pump pulse. In DFWM, this is achieved with separate pump and Stokes pulses with different angle of incidence, enabling control over the coherence generation by modulating the time delay between them. Similarly, in IVS the VC signals are detected in the same direction as the probe pulses, that is, self-heterodyne detection. By contrast, in DFWM, the BOXCARS geometry means the VC signals can solely be detected in the opposite direction of the probe pulses, that is, homodyne detection. Homodyne detection intrinsically shortens the oscillation decays and thereby broadens the FT spectra due to the result of heterodyning between scattering contributions from population gratings, but this limitation can be overcome by carrying out heterodyne detection with an external local oscillator.
Two-dimensional electronic spectroscopy (2DES) has also been applied as a time-domain Raman method [Citation74–76]. Performed in a BOXCARS four-wave-mixing geometry as in DFWM, the first two pulses in the excitation sequence generate VCs on both the ground and excited PESs. Unlike the other methods, the resulting signals are simultaneously resolved along excitation and detection frequency. The effects of VCs are detected as oscillations of the signal magnitude at a given excitation-detection point as a function of waiting time between the 2nd (k2) and 3rd (k3) pulses in the sequence, and thus Fourier transformation can again be applied to convert the time-domain data into the corresponding Raman signatures. The 2D detection scheme offers unique advantages for the analysis of VCs – whether they are on- or off-diagonal, how they shift as a function of beating frequency, whether they are favoured for parallel or orthogonal polarization of excitation and detection pulses [Citation77–79] – that can afford clear evidence of the ground- versus excited-state origin of the signals ())[Citation80].
The same concepts that underpin these time-domain techniques starting from the ground electronic state apply equally well to upper excited states. Thus, if an excited-state population is already prepared by an actinic pump, a broadband impulsive excitation pulse can be tuned to be solely resonant with SE or ESA regions to selectively generate excited-state VC. The corresponding experiments are known as Pump-IVS/Time-Resolved ISRS (TR-ISRS) and Pump-DFWM, and they provide new flexibility in the use of VCs to track structural dynamics [Citation63,Citation64,Citation69,Citation81–84]. Standard IVS or DFWM schemes only allow observation of VCs generated through the initial impulsive pump, and these dephase on the timescale of the vibrational-free induction decay (~ps). These techniques are therefore blind to structural changes that occurs on longer timescales. With Pump-IVS and Pump-DFWM, the timing of VC generation is precisely controlled through the interval between actinic and impulsive pumps. This permits the measurement of excited-state Raman spectra at different delay points and thus real-time tracking of continuous structural evolution ())[Citation85].
Similar insight can be gained from other, frequency-domain ultrafast vibrational techniques such as time-resolved infrared spectroscopy or femtosecond stimulated Raman spectroscopy. Both allow the observation of ultrafast photoinduced reactions from the perspective of nuclear coordinates. However, in the context of SF these are most frequently applied analogously to transient absorption, where vibrational fingerprints are used to track the growth and decay of different electronic populations [Citation86–89]. Only time-domain Raman spectroscopy provides the unique ability to watch coherent nuclear wavepacket motions in real time on the excited-state PES over the course of the reaction. Analysis of wavepacket dynamics in the many examples reported to date – for example, classical femtochemistry in small diatomic molecules [Citation90,Citation91], vision [Citation92–97], photosynthesis [Citation5,Citation75,Citation80,Citation98,Citation99], proton transfer in biological systems [Citation100–102], charge generation dynamics in organic and inorganic materials [Citation103–107] – has provided deep new insight into the role of vibronic coupling for ultrafast reactions in the non-adiabatic (non-Born-Oppenheimer) regime. Technical advances in recent years have significantly lowered the entry barrier to apply time-domain Raman spectroscopy. Improvements in the experimental and analytical approaches mentioned above have provided robust means to isolate the analytically meaningful excited-state VCs. At the same time, it has now become easier than ever to achieve generate stable broadband pulses and compress them towards the transform limit, the essential prerequisite to observe tiny VC signals on top of large electronic contributions [Citation63]. Thanks to these advances, time-domain Raman spectroscopy has recently served as a decisive proof of principle tool for vibronic coupling mechanisms in ultrafast 1TT formation, which we address in detail in the following section.
3. Vibronic coupling mechanisms for coherent triplet-pair state formation
Since the ground-breaking report of quantum coherent SF in pentacene films in 2011 [Citation42], there has been considerable interest in the detailed mechanism of the ultrafast conversion S1 → 1TT. Time-domain Raman techniques are well suited to this problem, given their intrinsically fast time resolution and the ability – in principle – to correlate the electronic dynamics with relevant nuclear motions, laying the basis for a fully resolved ‘molecular movie’ of the process. Accordingly, a range of ultrafast time-domain Raman techniques have been applied to multiple well-established SF systems, highlighting how coherent 1TT formation is activated by vibronic coupling mechanisms. This general mechanistic behaviour has been approached from multiple perspectives, and we group the principal studies below into thematic subsections: 1) Conical intersection dynamics, 2) Vibronically mixed S1-1TT manifolds, and 3) Observation of symmetry-breaking vibrations. In the fourth subsection, we briefly review related findings on vibronic coupling mechanisms of SF studied by other experimental approaches.
3.1 Conical intersection dynamics
Conical intersections (CIs) are widely recognized as gateways for ultrafast relaxation dynamics in a host of photochemical and photobiological reactions [Citation108–110]. Charge recombination and internal conversion in organic materials [Citation111–115], photoisomerization of retinal or molecular rotors [Citation1,Citation116–119], and protection of DNA against UV-induced photodamage [Citation120,Citation121] are but a few examples where CIs are thought to play a pivotal role. In general, a CI is the point or seam where two multidimensional potential energy surfaces (PESs) intersect, meaning separate treatment of electronic and nuclear coordinates is impossible. The simplest such picture, a symmetry-allowed CI, requires the concerted involvement of totally symmetric and non-totally symmetric vibrations. The former, called tuning modes, drive wavepackets from the initial Franck-Condon region to the CI by bringing the electronic states into resonance. The latter are required to connect the two PESs of different symmetry and are called coupling modes. The strong interactions between electronic and nuclear motions (vibronic coupling) at the CI enable non-adiabatic population transfer between different PESs.
Whereas a vast set of theoretical works suggest the involvement of CIs in ultrafast-photoinduced reactions [Citation122,Citation123], directly observing such level crossings between reactant and product states has proved experimentally demanding – often the presence of a CI is simply inferred from the high rate of reaction [Citation124,Citation125]. More challenging still is to determine the importance of these CIs in ultrafast processes. One method to more directly interrogate the involvement of CIs is by tracking the fate of VCs generated through impulsive photoexcitation: unlike other mechanisms to convert electronic states, a coherent nuclear wavepacket should survive passage through a CI (). Pump-probe-based IVS is the simplest technique to achieve this goal, though it is crucial to fully disentangle excited-state VCs from spurious ground-state signatures (see Section 2 for experimental considerations).
Figure 4. (a) A schematic illustration of a conical intersection between S1 and 1TT PESs in SF dynamics. Adapted from [46], copyright 2015 Springer Nature. (b) Structures of molecular systems reported to exhibit conical intersection dynamics
![Figure 4. (a) A schematic illustration of a conical intersection between S1 and 1TT PESs in SF dynamics. Adapted from [46], copyright 2015 Springer Nature. (b) Structures of molecular systems reported to exhibit conical intersection dynamics](/cms/asset/c30915ed-8811-4386-9012-c81dc14291c8/tapx_a_1918022_f0004_oc.jpg)
In 2015, Musser et al. first applied this concept to understand the mechanism of ultrafast SF, focusing on polycrystalline films of TIPS-pentacene ()) where 1TT is formed in quantitative yield [Citation46]. Here, the close overlap of the impulsive pump (500–650 nm, 10 fs) and the vibronic progression of the of S0 → S1 absorption transition enabled optimal generation of Franck-Condon active VCs on the S1 PES. The resulting pump-probe spectra reflected the formation of 1TT with an ~80 fs time constant, as well as distinct oscillations throughout the detection bandwidth due to VCs. The corresponding wavelength-resolved impulsive Raman map revealed complex, overlapping contributions from Raman signatures of ground and excited electronic states (), top). However, the use of an additional population-control pulse selective for 1TT allowed isolation of pure excited-state VCs (), bottom). Strong vibrational signatures were then only observed within the 1TT absorption bands, despite the original VC being generated through impulsive pumping of the S1 state. Such a surprising result could only be explained through coherent transfer of the impulsively generated S1 VCs to the 1TT PES, which entails the existence of a CI between them – the first report of a multimolecular CI. This first experimental evidence of CIs in ultrafast 1TT formation became a stepping stone for the generalization of the vibrationally coherent SF mechanism. However, the SF reaction was so fast as to preclude any observation of the initial S1 VC, and the product 1TT VC was found to roughly match that state’s intrinsic vibrational spectrum. As such, no determination was made about the role of the observed vibrations as tuning or coupling modes, or even as spectators which simply report the transfer process. Subsequent theoretical work from Petelenz and colleagues on the origins of these signatures was not able to shed light on this question but did confirm that the VCs must be transferred from S1 and are not simply generated in 1TT a product of the SF reaction [Citation126].
Figure 5. (a) Wavelength-resolved impulsive Raman map of a TIPS-pentacene film obtained from two-pulse, pump-probe, experiment (top) and three-pulse, pump-dump-probe, experiment (bottom). As can be seen in the bottom panel, a dump pulse controls the excited-state population so that the subtraction of VCs in the GSB region could be effectively conducted. Adapted from [46], copyright 2015 Springer Nature. (b) Wavelength-resolved impulsive Raman map of a DP-Mes thin film obtained by the two-pulse IVS experiment. (c) Simulation results showing projected total time-dependent mode displacements for all vibrations classified by their symmetry. Displacement amplitudes for A2, B1 and B2 modes are ~200-fold smaller than for A1 modes. (d) Comparison between experimental and calculated impulsive Raman spectra in the case of S1 → 1TT coherence transfer (top) and direct coherence generation on the 1TT PES (bottom). Adapted from [127], licensed under a Creative Commons Attribution 4.0 International License
![Figure 5. (a) Wavelength-resolved impulsive Raman map of a TIPS-pentacene film obtained from two-pulse, pump-probe, experiment (top) and three-pulse, pump-dump-probe, experiment (bottom). As can be seen in the bottom panel, a dump pulse controls the excited-state population so that the subtraction of VCs in the GSB region could be effectively conducted. Adapted from [46], copyright 2015 Springer Nature. (b) Wavelength-resolved impulsive Raman map of a DP-Mes thin film obtained by the two-pulse IVS experiment. (c) Simulation results showing projected total time-dependent mode displacements for all vibrations classified by their symmetry. Displacement amplitudes for A2, B1 and B2 modes are ~200-fold smaller than for A1 modes. (d) Comparison between experimental and calculated impulsive Raman spectra in the case of S1 → 1TT coherence transfer (top) and direct coherence generation on the 1TT PES (bottom). Adapted from [127], licensed under a Creative Commons Attribution 4.0 International License](/cms/asset/1fa77e61-2722-463f-a777-7c4f244284ae/tapx_a_1918022_f0005_oc.jpg)
These experimental limitations were addressed in a subsequent study applying the same pump-probe IVS method to DP-Mes [Citation127], which had been previously reported to undergo polarity-controlled sub-ps intramolecular SF [Citation128]. Despite the markedly slower timescale of SF in DP-Mes films (~320 fs) compared to TIPS-pentacene, transfer of VC from S1 to 1TT was again observed ()), reinforcing the generality of the CI-based mechanism for SF. More importantly, the authors discovered that the VC detected on 1TT following coherence transfer differed from the intrinsic Raman profile of 1TT, containing additional low-frequency modes. To understand this surprising effect, the authors applied a recently developed, fully quantum-mechanical tree-tensor network simulation (TTNS) approach [Citation129] incorporating all 108 atoms of DP-Mes, five electronic excited states 1TT and pairs of symmetry-adapted singlet and CT states, 252 vibrational modes spanning 110–1680 cm−1 and a linear vibronic Hamiltonian parameterizing their couplings to each state. In a departure from previous approaches the simulation was benchmarked not only by the experimental SF rate and (CT-mediated) pathway, but also by the frequencies and intensity profile of VC, reproducing both the phenomenon of coherence transfer and the appearance of ‘extra’ modes following coherence transfer through SF [Citation127]. Based on this high fidelity to experiment, the simulation could be treated as a detailed molecular movie of SF and enabled delineation of the nuclear motions into tuning modes (totally symmetric) and coupling modes (non-totally symmetric). The tuning modes were found to be displaced ~100x more than coupling modes ()), resulting in a ~ 10000x greater Raman intensity. Therefore, the observed VCs could only be assigned to tuning modes ()). This result sets a challenging bar for the direct observation of coupling modes, which should generally show extremely weak Raman intensity. The new vibrational modes detected in 1TT following SF were also classed as tuning modes, but they further serve as unique signatures of the passage of the vibrational wavepackets through a CI. The details of the molecular movie revealed the coordinated interplay of tuning modes – which dominate the early-time dynamics by modulating the inter-pentacene bond and induce S1-1TT resonance – and coupling modes – which subsequently activate and distort the symmetry of the molecule to drive population transfer.
This work powerfully highlights how state-of-the-art spectroscopic techniques and quantum calculations can be combined to achieve newly detailed insight into the real-time molecular motions that trigger SF and, indeed, many other ultrafast photochemical processes. Further theoretical analysis of the VCs by Andrzejak et al. raises an important cautionary note, though [Citation130]. The intensity profile and frequencies of VC detected in these experiments depend sensitively on the Franck-Condon factors of the transition through which VC is generated, the detailed overlap of this transition with the impulsive pump and the Franck-Condon factors of the excited-state transition in which VC is probed. These factors, which can be challenging to determine rigorously, must be accounted for in order to extract full atomistic detail from the observed VC response, underscoring the complexity of analysis in even this relatively simple experiment.
In 2020, Duan et al. employed a simpler conceptual scheme to qualitatively describe comparable CI-mediated SF dynamics in polycrystalline pentacene films [Citation45]. This work utilized a combination of 2DES and heterodyne-detected TG spectroscopy in one-colour experiments using ~18 fs pulses spanning 650–750 nm. Crucially, as a pump this pulse covered only the red edge of the pentacene absorption spectrum and thus principally favoured the generation of low-frequency and ground-state VC, while as a probe it enabled simultaneous tracking of the GSB, S1 SE and 1TT ESA. The chief result of the 2DES measurements was that, contrary to earlier ultrafast spectroscopy results on pentacene [Citation42], there is no evidence for significant electronic coherence driving SF. Instead, the TG spectroscopy revealed comparable VC signatures to those discussed above (albeit weighted towards much lower frequencies due to the pulse width of the pump), suggesting coherent population transfer from reactant to product PESs. The authors analysed the wavepacket dynamics of the VCs in low- and high-frequency regions with wavelet analysis to capture their time-domain evolution () and 6(b)), working on the assumption that all detected VCs reflected excited electronic population. The high-frequency VC near 1300 cm−1 dephased within 150 fs, roughly consistent with the SF timescale. At low frequencies, the initial 170 cm−1 VC detected in the GSB band split over ~500 fs into distinct modes at 140 and 250 cm−1. By contrast, the 140 cm−1 VC was detected in the 1TT ESA band from early times and exhibited no splitting or frequency shifts. The authors assigned the unusual peak splitting behaviour in the GSB and loss of high-frequency modes to changes of the vibrational frequencies after coherence transfer from the S1 (170 cm−1) to the 1TT (140 cm−1) states.
Figure 6. Oscillations due to VC and corresponding wavelet analysis for polycrystalline pentacene film in the (a) GSB and (b) ESA regions. Arrows highlight the splitting or persistence of low-frequency modes, while boxes indicate the short lifetime of high-frequency VC. (c) Calculated low-frequency (177 cm−1, left) coupling and high-frequency (1013 cm−1, right) tuning modes and constructed potential energy curves of the S1 and 1TT states along these coordinates. (d) Calculated wavepacket dynamics in the S1 (top) and 1TT (bottom) states along the coupling (left) and tuning mode coordinates of the CI, revealing VC transfer. Adapted from [45], licensed under a creative commons attribution 4.0 International License
![Figure 6. Oscillations due to VC and corresponding wavelet analysis for polycrystalline pentacene film in the (a) GSB and (b) ESA regions. Arrows highlight the splitting or persistence of low-frequency modes, while boxes indicate the short lifetime of high-frequency VC. (c) Calculated low-frequency (177 cm−1, left) coupling and high-frequency (1013 cm−1, right) tuning modes and constructed potential energy curves of the S1 and 1TT states along these coordinates. (d) Calculated wavepacket dynamics in the S1 (top) and 1TT (bottom) states along the coupling (left) and tuning mode coordinates of the CI, revealing VC transfer. Adapted from [45], licensed under a creative commons attribution 4.0 International License](/cms/asset/3b658948-704d-4f27-930e-1eeddc3fa820/tapx_a_1918022_f0006_oc.jpg)
Having identified some nuclear motions coordinated with the electronic dynamics, the authors applied a quantum mechanical-molecular mechanical approach to a model pentacene dimer pair to evaluate the role of each mode. They found large Huang-Rhys factors on the S1 and 1TT surfaces for both modes and a strong modulation of the S1-1TT electronic coupling along the low-frequency coordinate (), left). This prompted assignment as a coupling mode, despite the unexpectedly large Huang-Rhys factor for a mode that should be non-totally symmetric. The high-frequency mode was instead found to effectively modulate the energy gap between states (), right). Building on these mode assignments, the authors numerically simulated the wavepacket dynamics in a simple two-state (S1 and 1TT) two-mode (177 and 1013 cm−1) CI model ()). The model qualitatively reproduced the experimental SF timescale and phenomenon of VC transfer. However, the simulation revealed transfer of high-frequency and loss of low-frequency VC, opposite to what was experimentally reported. This is likely linked to the oversimplification of the complex vibronic dynamics into just two modes, but the absence of transferred coupling-mode VC is entirely consistent with the picture from Schnedermann et al [Citation127]. Importantly, the theoretical models in both studies provide a novel platform to directly explore the functional role of specific molecular motions, and both showed that artificially reducing the vibronic coupling strength of the coupling mode results in a significant slowdown in the rate of wavepacket transfer. The ultrafast dynamics of SF thus depend powerfully on the synchronization of these nuclear motions, even when the reaction is energetically favoured.
3.2 Vibronically mixed S1-TT manifolds
The perspective of SF as driven by a CI is most clearly formulated in terms of pure electronic states such as S1 and 1TT, the different symmetry properties of which prevent meaningful direct coupling between them. At the CI, the transfer of the wavepacket from one PES to another results in an instantaneous change in electronic state. In addition to this essentially diabatic picture, it is possible to formulate the SF process in terms of mixed adiabatic states, where evolution along certain nuclear coordinates – coupling to vibrations – results in a change in the balance of different diabatic contributions to the total wavefunction and thus a change in the character of the state from S1-like to 1TT-like () [Citation44].
Figure 7. A schematic illustration of vibronically mixed S1-1TT manifolds. Adapted from [131], copyright 2017 Springer nature. (b) Structures of molecular systems reported to exhibit vibronically mixed S1-1TT manifolds
![Figure 7. A schematic illustration of vibronically mixed S1-1TT manifolds. Adapted from [131], copyright 2017 Springer nature. (b) Structures of molecular systems reported to exhibit vibronically mixed S1-1TT manifolds](/cms/asset/3de2ac8d-d348-49a3-a2a4-85b093c15df9/tapx_a_1918022_f0007_oc.jpg)
This perspective naturally leads to the concept of vibronic mixing between S1 and 1TT, as first laid out experimentally in a ground-breaking 2DES study of polycrystalline pentacene, TIPS-pentacene and DT-pentacene thin films [Citation47]. Using 635–775 nm (14 fs) excitation pulses in a BOXCARS geometry, Bakulin et al. observed no significant signatures of electronic population following excitation at 1.72 eV, the assumed energy of the 1TT state – an expected result, due to the negligibly small transition dipole moment of 1TT. Instead, 1TT was only formed following excitation of higher-lying S1, in all cases with ~100 fs time constant, in accord with standard pump-probe observations. The impulsive excitation additionally resulted in long-lived oscillations of the entire 2D signal, due to VC (electronic coherence could be excluded by the absence of oscillations matching the frequency of the S1-1TT gap). Processing of this data into 2D beating maps yielded the surprising observation of high-frequency VCs (1170 and 1360 cm−1 in pentacene) detected at the 1TT excitation energy of 1.72 eV ()). Thus, despite the absence of electronic population in those conditions and the inability to directly photogenerate 1TT, the 2DES response showed distinct oscillations following excitation at its frequency ()).
Figure 8. (a) Experimental 2D beating maps for the 1170 cm−1 mode of pentacene. (b) Wave-mixing energy ladder diagrams for each marked circle in the beating maps. The |g>→|1TT> transitions are nominally forbidden but become weakly allowed through vibronic state mixing. (c) Comparison between experimental absorption spectra and calculated transitions from the ground state resulting from vibronic mixing between diabatic singlet and 1TT manifolds. Population kinetics for dark and bright states predicted by the vibronically mixed S1-1TT model reproduce experimental result. Adapted from [47], copyright 2016 Springer Nature
![Figure 8. (a) Experimental 2D beating maps for the 1170 cm−1 mode of pentacene. (b) Wave-mixing energy ladder diagrams for each marked circle in the beating maps. The |g>→|1TT> transitions are nominally forbidden but become weakly allowed through vibronic state mixing. (c) Comparison between experimental absorption spectra and calculated transitions from the ground state resulting from vibronic mixing between diabatic singlet and 1TT manifolds. Population kinetics for dark and bright states predicted by the vibronically mixed S1-1TT model reproduce experimental result. Adapted from [47], copyright 2016 Springer Nature](/cms/asset/a3291789-bb60-4844-9d37-8e1007d911ff/tapx_a_1918022_f0008_oc.jpg)
To explain this unusual behaviour, the authors developed a model of vibronic multiexciton manifolds, the core proposition of which is wavefunction mixing between S1 and the higher vibrational levels of 1TT when they approach resonance ()). Within this framework, the beating peaks observed from 1.72 eV excitation reflect a coherent superposition of 1TT and vibrationally excited 1TT’ states, which are vibronically mixed with S1 and thus partially optically allowed. The crucial feature is the involvement of appropriately high-frequency modes, like the tuning modes discussed in the previous section, to bring 1TT’ into resonance with S1. More broadly, the vibronic model of mixing between S1 and 1TT results in the formation of a dense manifold of 16 eigenstates with varying TT character. Among these, the lowest state exhibits 95% TT character but is not completely dark, explaining the oscillatory photoresponse. Dynamical simulations based on this model fully reproduced the experimental beating maps as well as the sub-100 fs conversion from bright S1 to dark 1TT states. As in simpler pump-probe IVS, the excellent agreement with the theoretical model firmly demonstrates the crucial role of vibronic coupling – here formulated in terms of state mixing – to drive the ultrafast formation of 1TT.
A similar picture emerged from subsequent investigation of SF in amorphous and polycrystalline thin films of the nominally endothermic system TIPS-tetracene, using the same broadband IVS methods applied to TIPS-pentacene and DP-Mes with 16 fs excitation pulses [Citation131]. Despite the marked overall endothermicity (E(S1) < 2E(T1), energy barrier ~180 meV), the concomitant decay of S1 SE and 1TT ESA in the pump-probe data revealed prompt SF with a surprisingly fast time constant of 250 fs. The basis for this behaviour lay in the excited-state VCs simultaneously detected in the same experiment. Two dominant modes were observed, at 315 and 760 cm−1, and from their intensity distribution with probe wavelength they could be assigned uniquely to S1 and 1TT, respectively. The authors analysed the temporal evolution of the VC frequency contents using a sliding-window Fourier transform with 1 ps window, which can provide similar information to the wavelet analysis used by Duan et al [Citation45]. In TIPS-tetracene, this analysis showed that on early times (0–1 ps) the 315 cm−1 S1 VC is prominently observed. Over the ensuing 400 fs, the intensity of this mode continually decreases while that of the 760 cm−1 1TT mode gradually rises. This dynamic demonstrates that the VC in 1TT is not directly photogenerated but instead forms as a product mode of the reaction. The implication is that nuclear relaxation within S1 facilitates rapid movement of the system away from the Franck-Condon region and disproportionately stabilizes the 1TT state to near-resonance. Once again, this model hinges on strong vibronic coupling – evidenced here through the transient VC signatures – to modify the energies of and couplings between S1 and 1TT and produce mixed states. The broader picture that results is that such vibronic coupling can enable the stabilization of the 1TT state well below the energy 2E(T1), allowing ultrafast SF to proceed even in endothermic systems through the same basic mechanism as in exothermic pentacenes.
A recent polarization-dependent 2DES study by Wang et al. likewise pinpoints coherent S1-1TT vibronic mixing as the cause of ultrafast 1TT formation in endothermic tetracene single crystals [Citation79]. The authors observed surprisingly fast sub-picosecond 1TT population kinetics, with a distinct excitation-wavelength dependence that suggested important involvement of vibrationally hot S1ʹ states and/or the upper Davydov S1* state. Because the 1TT state of tetracene is located above S1, the 2D beating maps revealed VCs only from the ground state; in contrast to pentacene [Citation47], the impulsive pump could not span the vibronic structure of the (dark) S0→1TT transition. Analysis of the high-frequency beating maps revealed strong peaks at energies intermediate between the lower S1 and upper S1* Davydov states, confirming the specific involvement of vibrationally hot S1ʹ. These features were ascribed to energetic resonance between the lower Davydov state and 1TT and corresponding vibronic interactions, contrary to the typically reduced vibronic activity of the J-type lower state.
3.3 Observation of symmetry-breaking vibrations
We specifically address in this section IVS studies of SF in rubrene ()). As formulated in the quantum dynamics work of Tamura et al., the crystal structure of rubrene is ideally suited to a vibronic mechanism [Citation132]. At the equilibrium geometry, the C2h symmetry results in strictly zero coupling between S1 and 1TT, even accounting for mediating CT states. During symmetry-breaking vibrations this is no longer the case, and substantially increased transfer integrals should provide large ‘direct’ coupling between these states to enable SF (). The authors thus predicted a CI between S1 and 1TT, with the symmetry-breaking vibrations playing the role of the non-totally symmetric coupling modes discussed in Sections 2 and 3.1. We note that a qualitatively similar picture applies to the orthogonal acene dimers DP-Mes [Citation127,Citation133] (Section 3.1) and DT-Mes [Citation134] (Section 3.4). In rubrene, it was suggested that slight thermal activation of the coupling coordinate would be sufficient to enable SF within the Franck-Condon region, yielding a temperature dependence in qualitative agreement with earlier spectroscopy on crystalline rubrene [Citation135]. As in the IVS study of DP-Mes above [Citation127], the displacements along non-totally symmetric coordinates are expected to be small, which is reflected by their small Huang-Rhys factors, and therefore their observation and assignment as coupling modes using time-domain Raman methods pose an extreme challenge.
Figure 9. (a) Molecular structure of rubrene. (b) A schematic illustration of the role of non-totally symmetric (symmetry-breaking) vibration in orbital overlaps between the HOMO and LUMO of adjacent molecules in symmetric rubrene single crystals. In perfect C2h symmetry (left), the coupling strength between S1 and 1TT (VSF) is close to zero because of the negligible transfer integrals. In contrast, when asymmetric torsion breaks the symmetry of the stacking geometry (right), it brings out non-zero VSF, enabling coherent SF. Adapted from [136], copyright 2017 Springer Nature
![Figure 9. (a) Molecular structure of rubrene. (b) A schematic illustration of the role of non-totally symmetric (symmetry-breaking) vibration in orbital overlaps between the HOMO and LUMO of adjacent molecules in symmetric rubrene single crystals. In perfect C2h symmetry (left), the coupling strength between S1 and 1TT (VSF) is close to zero because of the negligible transfer integrals. In contrast, when asymmetric torsion breaks the symmetry of the stacking geometry (right), it brings out non-zero VSF, enabling coherent SF. Adapted from [136], copyright 2017 Springer Nature](/cms/asset/055aa27b-d617-4624-8e52-6ceb00974ce8/tapx_a_1918022_f0009_oc.jpg)
Nevertheless, Miyata et al. reported the direct observation of symmetry-breaking vibrations enabling coherent 1TT formation in endothermic rubrene single crystals using IVS [Citation136]. In the electronic transient absorption data, the authors identified the characteristic 1TT ESA with two distinct rise components: a faster, temperature-independent component comparable to the instrument response of the system (~40 fs), and a slower, temperature-dependent component ~25 ps. These were attributed to coherent and incoherent 1TT formation, respectively. In the transient data measured at 35 K, the authors observed low-frequency VCs (<140 cm−1, limited by the time resolution of the pump) and analysed them based on the wavelength-resolved impulsive Raman map ()). Two dominant vibrations were revealed, denoted as mode α ~ 80 cm−1 and mode β ~ 124 cm−1. Whereas mode α was observed both in the 1TT ESA regions (480–550 nm and 750–850 nm) and the S1 ESA region (~620 nm), mode β appeared only in the 1TT ESA region. Based only on their probe spectral profile, these vibrational signatures were assigned to VC in S0 (α and β), S1 (only α) and 1TT (α and β).
Figure 10. (a) Wavelength-resolved impulsive Raman map of rubrene single crystal. (b) Calculated totally symmetric (top) and non-totally symmetric (bottom) modes, assigned as candidates for modes α and β indicated in panel a. (c) Calculated potential energy curves of the S0, S1, and 1TT states showing a CI for slight displacement along the SF coordinate. (d) A schematic illustration of how coherent curve crossing between S1 and 1TT is driven by the symmetry-breaking vibration (mode β) within the Franck-Condon region. Adapted from [136], copyright 2017 Springer Nature
![Figure 10. (a) Wavelength-resolved impulsive Raman map of rubrene single crystal. (b) Calculated totally symmetric (top) and non-totally symmetric (bottom) modes, assigned as candidates for modes α and β indicated in panel a. (c) Calculated potential energy curves of the S0, S1, and 1TT states showing a CI for slight displacement along the SF coordinate. (d) A schematic illustration of how coherent curve crossing between S1 and 1TT is driven by the symmetry-breaking vibration (mode β) within the Franck-Condon region. Adapted from [136], copyright 2017 Springer Nature](/cms/asset/baae076b-f6fc-4b6b-9bdb-e5014b42e4b0/tapx_a_1918022_f0010_oc.jpg)
On the basis of quantum mechanical-molecular mechanical calculations, the authors proposed that an in-phase seesaw vibration at 74 cm−1 could be responsible for mode α (), top), which is strongly Franck-Condon active with the S0 → S1 transition. Thus, it could be safely assigned to the totally symmetric Ag mode which cannot break the symmetry of the crystal. Mode α observed in the 1TT ESA region was ascribed to the result of a partial transfer of wavepackets from S1 to 1TT, akin to coherence transfer phenomena of tuning modes through a CI observed in pentacene derivatives. As for mode β in the 1TT ESA region, the authors assigned it to a symmetry-breaking mode that facilitates mixing the bright S1 and the dark 1TT within the Franck-Condon region. The calculation results suggested two candidates for mode β: an anti-phase Bg symmetry (117 cm−1) or in-phase Au symmetry (126 cm−1) backbone twist (), bottom).
The appearance of non-totally symmetric vibrations is an unusual phenomenon in impulsive Raman experiments and required further explanation. The authors suggested the key factor was that the experiments were performed with pump and probe polarized along the crystallographic b-axis, perpendicular to the a-axis orientation of the S0 → S1 transition, because of which the most prominent totally symmetric mode at 105 cm−1 was entirely absent. Moreover, the relative intensity between 80 cm−1 and 124 cm−1 modes in the impulsive Raman spectra deviated substantially from previously reported Raman spectra, implying possible contributions from non-Condon active vibrations. Based on these grounds to assign mode β to a coupling mode, the authors developed a model of a CI between S1 and 1TT PESs, located near the Franck-Condon region ()). In this region there is significant coupling and thus mixing between the states, resulting in coherent direct excitation of 1TT modulated by the antisymmetric mode. This ‘symmetry-breaking mode’ picture highlights through yet another formulation the crucial role nuclear motions can play to enable ultrafast SF, driving some population into coherent channels even in endothermic systems ()).
While these results are in qualitative accord with the quantum dynamics calculations of Tamura et al [Citation137]. – namely the existence of a CI between S1 and 1TT – they diverge in two crucial details. The CI was originally predicted to become less accessible at reduced temperature, whereas Miyata et al. only observed VC transfer at the lowest temperature of 35 K. Moreover, Tamura et al. predicted that phase interference should destroy the coherence of the initial wavepacket, such that SF through this CI should be an incoherent process, making the observation of excited-state VCs in rubrene especially surprising. Interestingly, very similar transient absorption experiments on a rubrene single crystal were reported by Breen et al., again polarized along the b-axis [Citation138]. This study concluded that ~20% of the initial S1 population transferred to 1TT within 25 fs in an ’incoherent’ fashion with the remainder undergoing SF on longer timescales. The authors’ experiments were based on 5.2 fs broadband visible pump and probe pulses spanning 470–750 nm, meeting the same partially resonant condition as Miyata et al. Surprisingly, no coherent oscillations were observed within the intense 1TT ESA or at any other detection wavelength, at odds with the results of Miyata et al [Citation136]. In 2DES measurements using the same excitation pulses, the authors likewise reported no VCs. The absence of observable S1-1TT vibronic state mixing effects [Citation47] can be explained by insufficient overlap of the impulsive pump with the dark S0→1TT transition, but the lack of VC in the IVS configuration is more challenging to reconcile. The authors interpreted this behaviour to show that VC generated in the S1 state does not transfer to the 1TT state, implying the absence of coherent vibronic coupling mechanisms for ultrafast 1TT formation in the rubrene crystal. Instead, on the basis of the evolving vibronic peak ratio within the 1TT ESA, a signature of vibrational relaxation, the authors suggested that the most important prerequisite for ultrafast SF in rubrene is energetic resonance between S1 and 1TT states or their higher vibronic levels. Such resonance can facilitate ultrafast equilibrium despite weak coupling between the two states; the weak coupling stems from librational disorder in the crystal present at any finite temperature, which breaks the local symmetry and is effectively static on the timescale of SF. The resonant coupling element of this model has conceptual similarity to the vibronic mixing picture of Bakulin et al [Citation47], albeit with lower overall coupling strengths that preclude coherent 1TT formation.
As such, while the importance of nuclear motion in ultrafast SF in crystalline rubrene is clear, it is challenging to assess the details of the vibronic coupling mechanism. A critical distinction which may explain the disparity between the two experimental studies is temperature, with the IVS results reported at 35 K and ultrafast incoherent SF reported at room temperature. The significant temperature effect on the dephasing time of VCs may materially alter the conclusions about the vibronic mechanism. Moreover, the limited overlap between impulsive pump and the absorption spectrum in both studies severely restricts the possibility to observe VC. Further experiments will likely be required with more resonant excitation to maximize the visibility of any excited-state VC. This will have the additional effect of favouring direct VC generation in 1TT through the 2DES scheme[Citation47], due to the higher relative 1TT energy in rubrene than the pentacenes.
3.4 Vibronic coupling mechanisms studied by other experimental approaches
The studies discussed above demonstrate how VC can be exploited as a tool to explore the central role of nuclear motion in ultrafast 1TT formation. Ultrafast time-domain Raman spectroscopy provides the most direct and intuitive techniques to ascertain the dynamical importance of vibronic coupling. However, these challenging experiments are not the only means to this end. A number of other studies likewise suggest SF is underpinned by vibronic coupling mechanisms, with noteworthy examples based on synthetic control, mixed S1-1TT transient absorption features, excitation wavelength-dependent SF and anomalous delayed fluorescence.
Indeed, one of the most straightforward approaches to investigate the role of nuclear motion is to constrain the vibrational landscape synthetically. In a set of pentacene dimers (BP series, ), Fuemmeler et al. incorporated different degrees of steric bulk at the bond governing the dihedral angle between pentacene cores [Citation139]. Transient absorption spectroscopy simply tracked the populations of S1 and 1TT states and revealed a reduction in the SF rate by a factor of 4.5 as the equilibrium dihedral angle increased from 37° to 57°. Theoretical analysis revealed that this degree of tuning was precisely in accord with predictions for a ‘direct’ mechanism of SF, one of the few clear such examples ()). The model further predicted that a high-frequency (1435 cm−1) ring-breathing mode with significant Huang-Rhys factor strongly modulated the relative energies of diabatic S1 and TT contributions, resulting in substantial changes in the character of the adiabatic states. Evolution along this coordinate results in an avoided crossing at resonance between S1 and TT, enabling ultrafast population transfer despite extremely small electronic coupling between the states. Thus, the 1435 cm−1 vibration plays the essential role of a tuning mode, similar to its role in coherent SF in the pentacene films above [Citation45–47].
Figure 11. Structures of molecular systems for which vibronic coupling mechanisms of SF have been reported through other experimental approaches
Figure 12. (a) Vibronic coupling driven intramolecular SF in BP series. Calculated potential energy curves along the 1435 cm−1 mode coordinate (upper left) and contributions of LE and TT diabatic states in the three lowest singlet adiabatic states (upper right). Adapted from [138], copyright 2016 American Chemical Society. (b) 2DES spectra of XanTDI2 in 1,4-dioxane at waiting time of 10 ps (left) and two schematic 2DES spectra on the basis of the superposition model (middle) and the equilibrium model (right). Spectral shape of experimental 2DES result follows the superposition model, indicating that the excitation-wavelength-dependent behaviour originates from the vibronic coupling. Adapted from [141], copyright 2018 American Chemical Society. (c) Solvent-polarity-dependent TT and CT action spectra of DT-Mes, which indicates the excitation-wavelength dependence of yields of the stabilized CT (CT↓) and 1TT states. Shaded spectra correspond to steady-state absorption and CT↓ emission. Dashed spectra indicate estimated distribution of the destabilized CT (CT↑) state. Adapted from [134], copyright 2019 American Chemical Society
![Figure 12. (a) Vibronic coupling driven intramolecular SF in BP series. Calculated potential energy curves along the 1435 cm−1 mode coordinate (upper left) and contributions of LE and TT diabatic states in the three lowest singlet adiabatic states (upper right). Adapted from [138], copyright 2016 American Chemical Society. (b) 2DES spectra of XanTDI2 in 1,4-dioxane at waiting time of 10 ps (left) and two schematic 2DES spectra on the basis of the superposition model (middle) and the equilibrium model (right). Spectral shape of experimental 2DES result follows the superposition model, indicating that the excitation-wavelength-dependent behaviour originates from the vibronic coupling. Adapted from [141], copyright 2018 American Chemical Society. (c) Solvent-polarity-dependent TT and CT action spectra of DT-Mes, which indicates the excitation-wavelength dependence of yields of the stabilized CT (CT↓) and 1TT states. Shaded spectra correspond to steady-state absorption and CT↓ emission. Dashed spectra indicate estimated distribution of the destabilized CT (CT↑) state. Adapted from [134], copyright 2019 American Chemical Society](/cms/asset/67a1df9b-3fa9-4f1e-8218-6e202ac4e42b/tapx_a_1918022_f0012_oc.jpg)
Other studies rely instead on the coexistence of S1 and 1TT features in transient absorption and other electronic spectroscopies, especially where they evolve on different timescales. Effects such as an increase in 1TT ESA without corresponding loss of S1 ESA are treated as a signature of the evolving character of the mixed S1-1TT wavefunction, which is most often described in terms of vibronic coupling. For instance, Monahan et al. applied time-resolved two-photon photoemission and transient absorption to polycrystalline thin films of highly exothermic hexacene () [Citation140]. The initial S1 state would found to decay with a lifetime of 180 fs, but the features of 1TT grew on the much faster timescale of 45 fs with subsequent 270 fs decay to form isolated triplets T1+ T1. The authors disentangled these complex dynamics with the aid of rate equation calculations and quantum dynamics simulations, finding that the major S1 decay channel is an incoherent SF pathway. However, the early-time 1TT formation could only be explained through coherent superposition of an excited vibrational level of 1TT with S1 due to near-resonance, in line with the model of vibronically mixed S1 and 1TT manifolds. Similarly, in TIPS-tetracene nanoparticles (), Thampi et al. observed a marked discrepancy between the timescales of S1 decay and 1TT formation, for example, 12 ps vs 2 ps following band-edge excitation, and it was suggested that this could reflect similar evolution of a vibronically mixed state [Citation141]. In addition, Thampi et al. found that the timescales of these processes were highly sensitive to the excitation wavelength, with slower and less efficient 1TT formation following photoexcitation of higher vibrational levels of S1. These conditions even resulted in different 1TT transient absorption spectral shapes, including in the limiting case distinct 1TT stimulated emission through a Herzberg-Teller mechanism (see below). These findings suggest that the initial vibrational relaxation away from the Franck-Condon point along the mixed S1-1TT PES controls the SF pathway, even well beyond the initial 500 fs coherent SF regime studied by Stern et al [Citation131], revealing the significant effect of vibronic coupling into the incoherent regime.
The excitation dependence effects have been explored in greater detail in intramolecular SF systems. In the case of the slip-stacked terrylene diimide dimer XanTDI2 () [Citation142], this was performed using 2DES, which naturally lends itself to detailed investigation of the excitation axis. In the 2D spectra of XanTDI2, Mandal et al. identified three different spectral ranges carrying unique signatures of the S1, CT and 1TT states. At a waiting time of 10 ps, all state signatures were present regardless of the excitation wavelength (), left). However, the contribution of CT signals was enhanced at excitation into the 0–1 vibronic band, while the 1TT contribution was enhanced for excitation at the band edge, suggesting that higher-energy excitation alters the branching between 1TT and CT formation. Fitting to simple kinetic models, the authors determined that the most likely explanation for this variation was the adiabatic representation, describing a superposition of eigenstates mixed with different diabatic S1, CT, and 1TT characters (), right). These can mix effectively due to their close energetic proximity, and the authors inferred a vibronic mechanism from the excitation wavelength dependence: as in Thampi et al [Citation141], excitation of additional vibrational quanta in the S1 PES resulted in pronounced changes in the nature of the final state.
Such interplay of S1, CT and 1TT states was investigated in greater detail, both experimentally and theoretically, by Alvertis et al. for the tetracene dimer DT-Mes () [Citation134]. In this system, the solvent-dependent dynamics of photoluminescence revealed that coupling to the inter-tetracene torsional mode results in vibronic mixing of CT and S1 to enable CT emission. In a systematic investigation of the excited-state dynamics in eight solvents at 10 excitation energies, the authors revealed SF to be ‘activated’, meaning the process occurs only upon excitation distinctly above the band edge of S1, implying a need for excess vibrational quanta to enable effective S1-1TT coupling (mediated by CT). SF was only detected above a certain threshold, but the energy and shape of the excitation-wavelength threshold varied with solvent polarity, highlighting a complex interplay of vibrational excitation and tuneable CT states ()). The full behaviour could be rationalized with a simple model Hamiltonian in which the intra-tetracene torsional mode serves as the tuning coordinate. As in DP-Mes and rubrene [Citation127,Citation136], the ground-state symmetry results in zero coupling between S1 or CT and 1TT, meaning a further symmetry-breaking interaction is required. In low- or high-polarity solvents, this is supplied by symmetry-breaking vibrations, resulting in an avoided crossing between S1-CT and TT surfaces and incoherent SF. When the external solvent environment provides more substantial symmetry breaking and shifts the CT state into closer resonance with 1TT, coupling to the torsional coordinate provides significant vibronic mixing between S1, CT and 1TT states and a substantially faster coherent SF pathway.
The concept of symmetry breaking also crucially underpins the final class of vibronic SF mechanisms. In a broad range of SF materials, spanning acenes [Citation15,Citation131,Citation143], heteroacenes [Citation15], zethrenes [Citation144], diketopyrrolopyrroles [Citation145], perylene diimides [Citation146], and conjugated polymers [Citation147,Citation148], with SF energetics ranging from highly endo- [Citation131,Citation141] to highly exothermic [Citation143], careful correlation of transient absorption and transient photoluminescence spectroscopy reveals unique red-shifted photoluminescence signatures that coincide with the dynamics of the 1TT state. While such observations have proved controversial in solution-phase measurements [Citation149,Citation150], where competing assignments to excimers are challenging to rule out, new experiments in single-crystalline pentacene confirm the origin of this emission as the 1TT state [Citation143]. This phenomenon is now sufficiently widespread to suggest that the ability of 1TT to emit photons is a general characteristic, though the studies discussed above agree it should be a nominally forbidden process. The mechanism of this delated 1TT emission was elucidated by Yong et al. for the case of TIPS-pentacene and an anthradithiophene [Citation15]. Namely, distortion of the equilibrium intermolecular geometry – a symmetry-breaking vibration – enables 1TT to mix with and thus borrow intensity from the bright S1 state [Citation15]. This is equivalent to the Herzberg-Teller mechanism by which the symmetry-forbidden S1 states of carotenoids and other polyenes emit photons [Citation151,Citation152]. More importantly, it is precisely the counterpart of the vibronic mechanisms of S1→1TT formation discussed above, and yet another piece of evidence that S1 and 1TT are strongly connected by non-totally symmetric vibrations.
4. Discussion
While the details of the interpretation may differ, the studies reviewed above agree on one essential point: the energies of electronic states and couplings between them evolve dynamically throughout the process of SF, and this evolution is governed by nuclear motions. Time-domain Raman spectroscopy has enabled tracking coherent nuclear motions on the excited-state PES in real time, revealing that the rate and efficiency of SF is in many cases driven by vibronic coupling. We stress that the VCs exploited in the time-domain techniques above are a powerful observational tool but are not necessary for the photophysical mechanism. The broad bandwidths of coherent excitation are required to observe VCs, but the underlying precise coordination of electronic and vibrational dynamics implied by VC transfer or vibronic state mixing are entirely insensitive to the excitation condition. Hence, the works presented in Section 3.4 can arrive at similar conclusions without recourse to VC techniques. Taken together, this body of work demonstrates conclusively that it is insufficient to consider static states and their couplings to understand SF, particularly in the ultrafast regime.
This broad accord notwithstanding, there remain key differences between these studies that raise important questions. One major issue is coupling modes within a CI mechanism and whether they can be robustly observed and identified. We recall that the coupling modes must be non-totally symmetric vibrations, effectively playing the role of a gear to compensate the symmetry mismatch between two PESs. Non-totally symmetric vibrations generally show near-zero Huang-Rhys factors, meaning that the displacements along these coordinates between the minima of the ground-state PES and of the optically excited PES are negligible. By contrast, the totally symmetric tuning modes, which drive population to the degeneracy point, can be highly displaced. While these vibrations work in concert in a CI mechanism, the coupling modes are more strictly limited and hold greater significance for mechanistic understanding and materials design, and thus there is considerable interest in pinpointing them experimentally. All works described in this review adhere to the same basic picture that one or more asymmetric coupling modes connect S1 and 1TT, but two opposing viewpoints exist regarding the experimental observability of such modes.
Schnedermann et al. argue that the coupling modes in DP-Mes cannot be detected in the impulsive Raman spectra because of their vastly smaller intrinsic displacement relative to the totally symmetric tuning modes [Citation127]. This interpretation is in line with the description above and was suggested to be a general challenge for vibrational spectroscopies seeking to describe the fine details of non-Born-Oppenheimer dynamics. The proposed solution – to stringently benchmark high-level simulations based on the VC behaviour of coupling modes – provides some guidance about the coupling modes but remains semi-quantitative. Having a stark difference between coupling and tuning mode displacement corresponds to the case of the A-term in Albrecht’s resonance Raman theory [Citation153], where the Raman enhancement factor is governed by the transition dipole moment and vibrational overlap integrals, that is, a Franck-Condon factor. In the DP-Mes experiment, the full overlap of the strongly allowed S1 absorption and the impulsive pump pulse satisfied the conditions of A-term enhancement. On the other hand, Miyata et al. claimed that the ‘symmetry-breaking’ coupling mode could be directly observed [Citation136]. The different experimental conditions for IVS on the rubrene single crystal – smaller overlap with the absorption and a markedly weaker S1 transition because of the b-axis polarization – mean that the Albrecht B-term enhancement was likely to have exerted a strong influence on the impulsive Raman spectra. The B-term describes the Herzberg-Teller vibronic coupling between the resonantly excited state and another state, if the energy gap between them is sufficiently small [Citation153]. In principle, totally and non-totally symmetric vibrations can both be enhanced through this term, and the wider consensus of significant vibronic coupling between S1 and 1TT suggests the assignment of the observed mode β as a symmetry-breaking non-Condon vibration is reasonable. Thus, these two perspectives on the observation of coupling modes through time-domain Raman spectroscopy have the potential to be reconciled, but further work is needed. Polarization-dependent IVS or the use of an array of different impulsive pumps providing different resonance overlap factors would provide invaluable insight into the variation of A- versus B-term contributions. Theoretical modelling including Duschinsky rotation and Herzberg–Teller coupling effects may be necessary to clarify this issue. While the latter may address the disagreement with the quantum dynamics simulations of Tamura et al[Citation137], which predicted no VC transfer, the contradictory result of Breen et al. leaves an open question [Citation138]. The controversial photophysics of crystalline rubrene are beyond the scope of this review [Citation154–158], but we note that the importance of symmetry breaking to enable SF in rubrene means that vibronic spectroscopies undoubtedly have an important role to play.
In addition to qualitative differences in the interpretation of coupling modes, the works presented above report quantitatively different VC spectra for the same state in the few instances where they measure the same material. In principle, some variation in VC is to be expected from variations in experimental conditions like resonance overlap. However, the most pronounced differences are reported in the polycrystalline pentacene films studied by Bakulin et al and Duan et al. [Citation45,Citation47], although both works used impulsive pump pulses centred nearly identically at ~700-nm with ~15 fs time resolution. The most prominent band reported by Duan et al., at 460 cm−1 (not analysed in detail), did not appear in the measurements of Bakulin et al. This mode may potentially be the well-known 462 cm−1 band from the quartz substrate, analogous to background solvent bands observed in solution IVS experiments, which would call for further experiments (e.g. using a population-control pulse or measuring pure solvent/substrate signals) or detailed Fourier intensity/phase analysis [Citation57,Citation66] to avoid misinterpretation. Nor do the other, weaker modes highlighted by Duan et al. appear in the spectra of Bakulin et al. The clear mismatch between vibrational spectra demands closer analysis. The field would greatly benefit from systematic application of the different VC techniques (IVS, transient-grating DFWM, 2DES) to the same materials to establish a clearer baseline understanding of whether, how and why they yield different results.
Furthermore, in both experiments, the impulsive pump was red-detuned such that it only covered the 0–0 vibronic band of the ground-state absorption (S0 → S1). In principle, this condition precludes the formation of high-frequency VC in S1 suggested by Duan et al., and it instead favours generation of ground-state VC through the ISRS mechanism. Bakulin et al. recognized this issue and took advantage of the absence of high-frequency S1 VC in their interpretation to validate an alternative hypothesis, distinguishing ground-state VC from excited-state – and thus 1TT – VC using rephasing/non-rephasing beating maps analysis. Indeed, the expected pentacene 1TT energy ETT = ~1.72 eV (720 nm) means its 0–0 and 0–1 vibronic transitions could be covered by the impulsive pump, the requirement for high-frequency VC generation. This example highlights the critical importance of the pump overlap factor discussed above in the interpretation of time-domain vibrational signals.
In this context, it is instructive to compare the TIPS-pentacene measurements of Musser et al. and Bakulin et al. [Citation46,Citation47] While both works used photogenerated VC to elaborate a mechanism of vibronic coupling between S1 and 1TT, the differences in resonance condition result in VC of fundamentally different characters. In the pump-probe IVS study, the excellent overlap between pump and absorption resulted in efficient generation of high-frequency coherence in the initial S1 state, and it was specifically the S1-originated wavepacket that was detected in 1TT. By contrast, and just as with pentacene, the off-resonant pulse in the 2DES work was incapable of generating such VC within the S1 state of TIPS-pentacene. However, it exhibited optimal overlap with the (dark) S0→1TT transition and its purported vibronic replicas, resulting in direct VC generation in 1TT. The 1TT impulsive Raman spectra resulting from these conditions should reflect the different Franck-Condon factors involved in their generation, namely S0→S1 (and any effects of the CI itself) versus S0→1TT. If they can be obtained with sufficient frequency resolution, and in combination with suitably high-level modelling, such a comparison should enable direct insight into the specific modes involved in the symmetry breaking that enables S1-1TT mixing. More broadly, the two studies reveal how VC can be exploited in conceptually different ways to uncover the same underlying physics of ultrafast SF driven by vibronic coupling.
Finally, we briefly point out the contributions to vibronic studies of SF made with frequency-domain vibrational methods, such as time-resolved infrared and picosecond/femtosecond stimulated Raman spectroscopy [Citation86,Citation88,Citation89,Citation159,Citation160]. Beyond their use to track the fingerprints and populations of electronic states [Citation86,Citation88], these measurements are more advantageous for tracking time-dependent changes in vibrational frequencies than those exploiting VCs. For instance, Deng et al. report the progressive blue-shift of a high-frequency mode in crystalline hexacene over precisely the same timescale as SF occurs [Citation159]. Using functional mode analysis, the authors assign this mode as the key reaction coordinate driving ultrafast exothermic SF. While wavelet analysis provides some analogous information about mode evolution in time-domain measurements, increased temporal resolution comes at a cost of reduced frequency resolution, limiting the ability of time-domain methods to detect subtle effects. The multi-pulse schemes Pump-IVS or Pump-DFWM capture similar information with high temporal resolution, but the VC signals must be constructed from time-domain oscillations spanning >1 ps, leaving them less sensitive to faster vibrational dynamics. The frequency-domain techniques thus provide valuable complementary insight into the evolution of the vibrational structure and have played a vital role in investigating the CT-mediated mechanism of SF, intermolecular dynamics and the vibrational signatures of 1TT [Citation89,Citation160]. A detailed description of these and related contributions can be found in the recent review by Frontiera and co-workers [Citation161].
5. Conclusion and outlook
The studies presented above demonstrate the importance of nuclear dynamics within SF. A range of time-domain Raman techniques have been applied to unravel the correlated electronic and nuclear motions that underpin 1TT formation in unprecedented detail, revealing the critical role of vibronic coupling between the initial and product states. It is especially striking how robust this behaviour is: VC can be maintained similarly in inter- and intramolecular materials; in exoergic, isoergic and endoergic systems; and with fission timescales spanning from 80 fs to nearly 1 ps. A central message from this body of work is the need to reframe how we think about SF – and, by extension, a host of other processes. It is conventional to describe photophysical dynamics in terms of simple ‘stick’ diagrams, representing pure electronic states of fixed energy and frozen geometry. The widespread observation of vibrationally coherent SF is a reminder that these stick diagrams, though convenient, are a gross simplification – the relevant dynamics occur on PESs defined by nuclear motion, where the states can significantly change in energy and even mix together. Full understanding of the fission dynamics thus requires an explicitly dynamical picture, and one of the key challenges is to bridge the gap between the static-states model that most naturally falls out of standard optical spectroscopy (photoluminescence, transient absorption) and the PESs that more complex techniques suggest ().
Figure 13. Implications of the vibronic mechanism of SF and prospects for further investigation
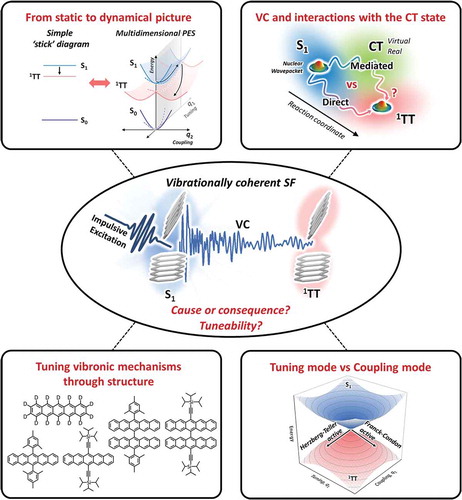
One important direction will be the further development of the relevant experimental tools. Two-dimensional Raman spectroscopy, for instance, can yield more detailed insight into the interactions between different nuclear coordinates [Citation112,Citation162,Citation163]. This would enable a transition from the simple detection of vibrationally coherent dynamics to an understanding of which motions couple together to drive 1TT formation. These techniques remain somewhat limited by their very complexity, both in execution and analysis, and it will be equally important to see commensurate developments in the modelling of complex vibronic dynamics. Recent advances using tree-tensor network states show that it is possible to efficiently compute the measured vibronic trajectories and supplement the experimental observables with atomistic detail [Citation127,Citation129]. It remains challenging, though, to incorporate the effects of finite temperature, external environment, delocalization (i.e. large system size) and disorder into such models, though all are likely to directly impact the observable dynamics. In an important step in this direction, Xue et al. performed ab initio molecular dynamics sampling on BODIPY dimers to understand an earlier report of surprisingly endothermic intramolecular SF [Citation164,Citation165]. The authors revealed that a combination of thermal motions on the ground-state potential energy surface and specific solvent–solute interactions – both capable of significantly modifying the state energies – was sufficient to overcome the energy barrier. In short, only through the vibrational energy landscape and the disorder associated with it could the fission behaviour be explained.
While these recent advances are promising, it is critical that new vibronic models of SF dynamics can be adequately benchmarked against experimental results. This requirement sets up yet another serious challenge. It is insufficient to simply compare calculated and observed SF rates: the long-lasting debate over ‘direct’ versus ‘mediated’ SF pathways in pentacene amply demonstrates how incompatible models can describe the same experimental kinetics. Although the classical rate equation models also consider vibrational degrees of freedom, for example by incorporating the Franck-Condon or Huang-Rhys factors, they are typically only treated in an averaged fashion. Generally, incoherent SF rates have been well described, indicating that in some regimes a detailed picture of vibronic dynamics is not needed. However, faster coherent dynamics cannot be fully explained by these models, as shown in the cases of crystalline hexacene, TIPS-tetracene or the tetracene dimer DT-Mes [Citation131,Citation134,Citation140]. Moreover, experimental [Citation45,Citation47] and theoretical [Citation49,Citation50,Citation52] work on crystalline pentacene has highlighted the importance of just a few specific vibrational modes in offsetting the energy mismatch between S1 and 1TT and vibronically coupling the two states: in some systems, it is evident that a general picture of the vibrational bath is insufficient. To better understand this behaviour, more detailed comparison is needed, as showcased in the benchmarking of TTNS simulations of SF in DP-Mes with the spectrum of transferred VC [Citation127]. Yet even in this state-of-the-art result, the remarkable agreement with the experimental data rests on a relatively small set of parameters governing the non-totally symmetric modes in the linear vibronic Hamiltonian. Thus, even the best methods cannot yet pinpoint the precise atomistic trajectory of complex dynamics like SF, but instead highlight the most probable and promising directions. More precise comparisons could be made by incorporating explicit consideration of the excited-state Franck-Condon factors and the details of the pulse spectral shape. As thoroughly demonstrated by Andrzejak et al. [Citation126,Citation130,Citation166], there can be no shortcuts here: slight approximations in the pump resonance conditions, molecular structure or Franck-Condon factors can result in erroneous predictions. Even so, the core problem remains that the structural data extracted from such optical experiments will only capture a limited portion of the phase space described by simulations. While we anticipate further improvement in this regard, it is likely the biggest breakthroughs will come from new spectroscopic techniques using ultrafast x-ray absorption, free-electron lasers or femtosecond electron diffraction that can directly monitor structural dynamics in real time following photoexcitation [Citation167–170]. Such methods would offer an entirely new level of insight into the correlated electronic and nuclear dynamics and singlet fission and provide even more rigorous validation of vibronic models.
A great deal remains to be explored, though, within the scope of methods currently available, which have only begun to be applied to complex materials systems. An open and challenging question is whether and how VC can be used as a tool to extract deeper mechanistic insight into other aspects of the fission process. For instance, the concept of vibrationally coherent transfer of a wavepacket from the initial S1 state to a product 1TT state maps most readily onto a model of ‘direct’ coupling between S1 and 1TT. On the other hand, in the pentacene dimer DP-Mes, it was systematically established that SF is mediated by virtual CT states [Citation128], yet VC transfer is clearly observed [Citation127]. How should these coherent dynamics be impacted by the involvement of electronic states that are not directly populated, and do these techniques provide a unique experimental observable to distinguish between these two mechanisms? More broadly, while there are now ample observations of VC in SF, there have been no investigations of how the phenomenon may be tuned. Alteration of the vibrational landscape – for example, through deuteration [Citation96], tuning the environment or substituting side groups – should directly impact the potential energy landscape and thus the dynamics and coherence properties of fission, but this behaviour has yet to be examined (). Will such control afford further insights into the mechanism of SF and the structural factors that govern its rate, or is the observation of VC simply a binary?
Chemical approaches like these to understand the tuneability of VC effects and the mechanistic information they can provide touch on a more general challenge. Powerful experimental tools developed in recent years have enabled the direct identification of vibronic dynamics in a host of materials and processes, from SF to photoisomerization, internal conversion to donor-acceptor charge transfer. The ostensible goal of many of these investigations is to better understand the underlying mechanism, with the ultimate aim to parlay that understanding into new molecular designs with optimal photophysical rates. But the question is rarely addressed, do these observations have a unique functional meaning? Are coherent vibronic dynamics a distinct property that can be tuned and optimized, or instead an inevitable byproduct or conjugate of fast electronic dynamics in organic materials? Put another way, can an ultrafast SF system be found which unambiguously fails to transfer VC? For coherent vibronic interactions to become a useful design principle, it will be equally important to begin to establish how many vibrational modes effectively contribute to a specific reaction coordinate and how they can control the reaction rate. Do large numbers of vibrational degrees of freedom collectively participate in the reaction coordinate, as suggested in rate model pictures? Can just a few vibrational modes accelerate the reaction, i.e., can we precisely discriminate between spectator and promotor modes? Identifying such limiting cases will help inform whether and how the mechanistic insights into SF delivered by VC techniques can be parlayed into new, more efficient molecular designs.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Polli D, Altoè P, Weingart O, et al. Conical intersection dynamics of the primary photoisomerization event in vision. Nature. 2010;467:440–43.
- Roberts GM, Hadden DJ, Bergendahl LT, et al. Exploring quantum phenomena and vibrational control in σ* mediated photochemistry. Chem Sci. 2013;4:993–1001.
- Ashfold MNR, King GA, Murdock D, et al. Πσ* excited states in molecular photochemistry. Phys Chem Chem Phys. 2010;12:1218–1238.
- Scholes GD, Fleming GR, Chen LX, et al. Using coherence to enhance function in chemical and biophysical systems. Nature. 2017;543:647–656.
- Fuller FD, Pan J, Gelzinis A, et al. Vibronic coherence in oxygenic photosynthesis. Nat Chem. 2014;6:706–711.
- Rafiq S, Fu B, Kudisch B, et al. Interplay of vibrational wavepackets during an ultrafast electron transfer reaction. Nat Chem. 2021;13:70–76.
- Gelinas S, Rao A, Kumar A, et al. Ultrafast long-range charge separation in organic semiconductor photovoltaic diodes. Science. 2014;343:512–516.
- Srinivasan R, Feenstra JS, Park ST, et al. Dark structures in molecular radiationless transitions determined by ultrafast diffraction. Science. 2005;307:558–563.
- Delor M, Scattergood PA, Sazanovich IV, et al. Toward control of electron transfer in Donor-acceptor molecules by bond-specific infrared excitation. Science. 2014;346:1492–1495.
- Delor M, Keane T, Scattergood PA, et al. On the mechanism of vibrational control of Light-induced charge transfer in Donor–Bridge–Acceptor assemblies. Nat Chem. 2015;7:1–7.
- Delor M, Archer SA, Keane T, et al. Directing the path of light-induced electron transfer at a molecular fork using vibrational excitation. Nat Chem. 2017;9:1099–1104.
- Bakulin AA, Lovrincic R, Yu X, et al. Mode-selective vibrational modulation of charge transport in organic electronic devices. Nat Commun. 2015;6. DOI:https://doi.org/10.1038/ncomms8880.
- Merrifield RE, Avakian P, Groff RP. Fission of singlet excitons into pairs of triplet excitons in tetracene crystals. Chem Phys Lett. 1969;3:140–143.
- Smith MB, Michl J. Singlet fission. Chem Rev. 2010;110:6891–6936.
- Yong CK, Musser AJ, Bayliss SL, et al. The entangled triplet pair state in acene and heteroacene materials. Nat Commun. 2017;8:15953.
- Musser AJ, Clark J. Triplet-pair states in organic semiconductors. Annu Rev Phys Chem. 2019;70:323–351.
- Zhu T, Huang L. Exciton transport in singlet fission materials: a new hare and tortoise story. J. Phys. Chem. Lett. 2018;9:6502–6510.
- Weiss LR, Bayliss SL, Kraffert F, et al. triplet pairs in an organic semiconductor. Nat Phys. 2017;13:176–181.
- Tayebjee MJY, Sanders SN, Kumarasamy E, et al. Quintet multiexciton dynamics in singlet fission. Nat Phys. 2017;13:182–188.
- Pensack RD, Tilley AJ, Grieco C, et al. Striking the right balance of intermolecular coupling for high-efficiency singlet fission. Chem Sci. 2018;9:6240–6259.
- Chen M, Krzyaniak MD, Nelson JN, et al. Quintet-triplet mixing determines the fate of the multiexciton state produced by singlet fission in a terrylenediimide dimer at room temperature. Proc Natl Acad Sci. 2019;116:8178–8183.
- Hanna MC, Nozik AJ. Solar conversion efficiency of photovoltaic and photoelectrolysis cells with carrier multiplication absorbers. J Appl Phys. 2006;100:074510.
- Smith MB, Michl J. Recent advances in singlet fission. Annu Rev Phys Chem. 2013;64:361–386.
- Rao A, Friend RH. Harnessing singlet exciton fission to break the Shockley–queisser limit. Nat Rev Mater. 2017;2:17063.
- Lee J, Jadhav P, Reusswig PD, et al. Singlet exciton fission photovoltaics. Acc Chem Res. 2013;46:1300–1311.
- Miyata K, Conrad-Burton FS, Geyer FL, et al. Triplet pair states in singlet fission. Chem Rev. 2019;119:4261–4292.
- Kim H, Zimmerman PM. Coupled double triplet state in singlet fission. Phys Chem Chem Phys. 2018;20:30083–30094.
- Sanders SN, Pun AB, Parenti KR, et al. Understanding the bound Triplet-pair state in singlet fission. Chem. 2019;5:1988–2005.
- Casanova D. Theoretical modeling of singlet fission. Chem Rev. 2018;118:7164–7207.
- Krishnapriya KC, Musser AJ, Patil S. Molecular design strategies for efficient intramolecular singlet exciton fission. ACS Energy Lett. 2019;4:1.
- Korovina NV, Pompetti NF, Johnson JC. Lessons from intramolecular singlet fission with covalently bound chromophores. J Chem Phys. 2020;152:040904.
- Greyson EC, Vura-Weis J, Michl J, et al. Maximizing singlet fission in organic dimers: theoretical investigation of triplet yield in the regime of localized excitation and fast coherent electron transfer. J Phys Chem B. 2010;114:14168–14177.
- Berkelbach TC, Hybertsen MS, Reichman DR. Microscopic theory of singlet exciton fission. I. general formulation. J Chem Phys. 2013;138:114102.
- Beljonne D, Yamagata H, Brédas JL, et al. Charge-transfer excitations steer the davydov splitting and mediate singlet exciton fission in pentacene. Phys Rev Lett. 2013;110:226402.
- Feng X, Luzanov AV, Krylov AI. Fission of entangled spins: an electronic structure perspective. J Phys Chem Lett. 2013;4:3845–3852.
- Kolomeisky AB, Feng X, Krylov AI. A simple kinetic model for singlet fission: a role of electronic and entropic contributions to macroscopic rates. J Phys Chem C. 2014;118:5188–5195.
- Zimmerman PM, Zhang Z, Musgrave CB. Singlet fission in pentacene through multi-exciton quantum states. Nat Chem. 2010;2:648–652.
- Zimmerman PM, Musgrave CB, Head-Gordon M. A correlated electron view of singlet fission. Acc Chem Res. 2013;46:1339–1347.
- Renaud N, Sherratt PA, Ratner MA. Mapping the relation between stacking geometries and singlet fission yield in a class of organic crystals. J Phys Chem Lett. 2013;4:1065–1069.
- Chan W-L, Berkelbach TC, Provorse MR, et al. The quantum coherent mechanism for singlet fission: experiment and theory. Acc Chem Res. 2013;46:1321–1329.
- Alguire EC, Subotnik JE, Damrauer NH. Exploring Non-condon effects in a covalent tetracene dimer: how important are vibrations in determining the electronic coupling for singlet fission? J Phys Chem A. 2015;119:299–311.
- Chan W-L, Ligges M, Jailaubekov A, et al. Observing the multiexciton state in singlet fission and ensuing ultrafast multielectron transfer. Science. 2011;334:1541–1545.
- Chan W-L, Ligges M, Zhu X-Y. The energy barrier in singlet fission can be overcome through coherent coupling and entropic gain. Nat Chem. 2012;4:840–845.
- Yost SR, Lee J, Wilson MWB, et al. A transferable model for singlet-fission kinetics. Nat Chem. 2014;6:492–497.
- Duan HG, Jha A, Li X, et al. Intermolecular vibrations mediate ultrafast singlet fission. Sci Adv. 2020;6:38.
- Musser AJ, Liebel M, Schnedermann C, et al. Evidence for conical intersection dynamics mediating ultrafast singlet exciton fission. Nat Phys. 2015;11:352–357.
- Bakulin AA, Morgan SE, Kehoe TB, et al. Real-time observation of multiexcitonic states in ultrafast singlet fission using coherent 2D electronic spectroscopy. Nat Chem. 2016;8:16–23.
- Ito S, Nagami T, Nakano M. Density analysis of intra- and intermolecular vibronic couplings toward bath engineering for singlet fission. J Phys Chem Lett. 2015;6:4972–4977.
- Tempelaar R, Reichman DR. Vibronic exciton theory of singlet fission. II. Two-dimensional spectroscopic detection of the correlated triplet pair state. J Chem Phys. 2017;146:174704.
- Tempelaar R, Reichman DR. Vibronic exciton theory of singlet fission. I. linear absorption and the anatomy of the correlated triplet pair state. J Chem Phys. 2017;146:174703.
- Morrison AF, Herbert JM. Evidence for singlet fission driven by vibronic coherence in crystalline Tetracene. J Phys Chem Lett. 2017;8:1442–1448.
- Tempelaar R, Reichman DR. Vibronic exciton theory of singlet fission. iii. how vibronic coupling and thermodynamics promote rapid triplet generation in pentacene crystals. J Chem Phys. 2018;148:244701.
- Xie X, Santana-Bonilla A, Fang W, et al. Exciton–phonon interaction model for singlet fission in prototypical molecular crystals.J Chem Theory Comput. 2019;15:3721–3729.
- Buckup T, Leonard J. Multidimensional vibrational coherence spectroscopy. Top Curr Chem Collect. 2019. DOI:https://doi.org/10.1080/00268978300100671
- De Silvestri S, Fujimoto JG, Ippen EP, et al. Femtosecond Time-resolved measurements of optic phonon dephasing by impulsive stimulated Raman scattering in α-perylene crystal from 20 to 300 K. Chem Phys Lett. 1985;116:146–152.
- Dhar L, Rogers JA, Nelson KA. Time-resolved vibrational spectroscopy in the impulsive limit. Chem Rev. 1994;94:157–193.
- De Silvestri S, Cerullo G, Lanzani G. Coherent vibrational dynamics. Lanzani G, Cerullo G, De Silvestri S editors. Chapter title: 1. Time Domain Vibrational Spectroscopy: Principle and Experimental Tools. Boca Raton: CRC Press; 2007;35-40. doi: https://doi.org/10.1201/9781420017519
- Polli D, Lüer L, Cerullo G. High-time-resolution Pump-probe system with broadband detection for the study of Time-domain vibrational dynamics. Rev Sci Instrum. 2007;78:103108.
- Brazard J, Bizimana LA, Turner DB. Accurate convergence of Transient-absorption spectra using pulsed lasers. Rev Sci Instrum. 2015;86:053106.
- Gueye M, Nillon J, Crégut O, et al. Broadband UV-Vis vibrational coherence spectrometer based on a hollow fiber compressor. Rev Sci Instrum. 2016;87:093109.
- Grupp A, Budweg A, Fischer MP, et al. Broadly tunable ultrafast pump-probe system operating at Multi-KHz repetition rate. J Opt. 2018;20:014005.
- McClure SD, Turner DB, Arpin PC, et al. Coherent oscillations in the PC577 cryptophyte antenna occur in the excited electronic state. J Phys Chem B. 2014;118:1296–1308.
- Liebel M, Schnedermann C, Wende T, et al. Principles and applications of broadband impulsive vibrational spectroscopy. J Phys Chem A. 2015;119:9506–9517.
- Liebel M, Kukura P. Broad-band impulsive vibrational spectroscopy of excited electronic states in the time domain. J Phys Chem Lett. 2013;4:1358–1364.
- Fitzpatrick C, Odhner JH, Levis RJ. Spectral Signatures of Ground- and excited-state wavepacket interference after impulsive excitation. J Phys Chem A. 2020;124:6856–6866.
- Cina JA, Kovac PA, Jumper CC, et al. Ultrafast transient absorption revisited: phase-flips, spectral fingers, and other dynamical features. J Chem Phys. 2016;144:175102.
- Bardeen CJ, Wang Q, Shank CV. Selective excitation of vibrational wave packet motion using chirped pulses. Phys Rev Lett. 1995;75:3410–3413.
- Wand A, Kallush S, Shoshanim O, et al. Chirp effects on impulsive vibrational spectroscopy: a multimode perspective. Phys Chem Chem Phys. 2010;12:2149–2163.
- Wende T, Liebel M, Schnedermann C, et al. Population-controlled impulsive vibrational spectroscopy: background- and baseline-free raman spectroscopy of excited electronic states. J Phys Chem A. 2014;118:9976–9984.
- Joo T, Albrecht AC. Vibrational frequencies and dephasing times in excited electronic states by femtosecond time-resolved four-wave mixing. Chem Phys. 1993;173:17–26.
- Goodno GD, Dadusc G, Miller RJD. Ultrafast heterodyne-detected transient-grating spectroscopy using diffractive optics. J Opt Soc Am B. 1998;15:1791.
- Brown EJ, Zhang Q, Dantus M. Femtosecond Transient-grating techniques: population and coherence dynamics involving ground and excited states. J Chem Phys. 1999;110:5772–5788.
- Kraack JP, Motzkus M, Buckup T. Selective nonlinear response preparation using femtosecond spectrally resolved Four-wave-mixing. J Chem Phys. 2011;135:224505.
- Dean JC, Scholes GD. Coherence spectroscopy in the condensed phase: insights into molecular structure, environment, and interactions. Acc Chem Res. 2017;50:2746–2755.
- Jumper CC, Rafiq S, Wang S, et al. From coherent to vibronic light harvesting in photosynthesis. Curr Opin Chem Biol. 2018;47:39–46.
- Cao J, Cogdell RJ, Coker DF, et al. Quantum Biology Revisited. Sci. Adv. 2020;6:eaaz4888.
- Egorova D. Detection of dark states in two-dimensional electronic photon-echo signals via ground-state coherence. J Chem Phys. 2015;142:212452.
- Egorova D. Self-analysis of coherent oscillations in time-resolved optical signals. J Phys Chem A. 2014;118:10259–10267.
- Wang G, Zhang C, Liu Z, et al. Singlet fission dynamics in tetracene single crystals probed by polarization-dependent two-dimensional electronic spectroscopy. J Phys Chem A. 2020;124:10447–10456.
- Dean JC, Mirkovic T, Toa ZSD, et al. Vibronic enhancement of algae light harvesting. Chem. 2016;1:858–872.
- Motzkus M, Pedersen S, Zewail AH. Femtosecond real-time probing of reactions. 19. nonlinear (DFWM) techniques for probing transition states of uni- and bimolecular reactions. J Phys Chem. 1996;100:5620–5633.
- Hauer J, Buckup T, Motzkus M. Pump-degenerate four wave mixing as a technique for analyzing structural and electronic evolution: multidimensional time-resolved dynamics near a conical intersection. J Phys Chem A. 2007;111:10517–10529.
- Kuromochi H, Takeuchi S, Tahara T. Femtosecond time-resolved impulsive stimulated raman spectroscopy using sub-7-fs pulses: apparatus and applications. Rev Sci Instrum. 2016;87:043107.
- Kraack JP, Wand A, Buckup T, et al. Mapping multidimensional excited state dynamics using pump-impulsive-vibrational-spectroscopy and pump-degenerate-four-wave-mixing. Phys Chem Chem Phys. 2013;15:14487.
- Kim W, Kim T, Kang S, et al. Tracking structural evolution during Symmetry‐breaking charge separation in quadrupolar perylene bisimide with time‐resolved impulsive stimulated raman spectroscopy. Angew Chemie Int Ed. 2020;59:8571–8578.
- Wang C, Tauber MJ. High-yield singlet fission in a zeaxanthin aggregate observed by picosecond resonance raman spectroscopy. J Am Chem Soc. 2010;132:13988–13991.
- Chen M, Bae YJ, Mauck CM, et al. Singlet fission in covalent terrylenediimide dimers: probing the nature of the multiexciton state using femtosecond Mid-infrared spectroscopy. J Am Chem Soc. 2018;140:9184–9192.
- Grieco C, Kennehan ER, Rimshaw A, et al. Harnessing molecular vibrations to probe triplet dynamics during singlet fission. J Phys Chem Lett. 2017;8:5700–5706.
- Hart SM, Silva WR, Frontiera RR. Femtosecond stimulated raman evidence for charge-transfer character in pentacene singlet fission. Chem Sci. 2018;9:1242–1250.
- Rose TS, Rosker MJ, Zewail AH. Femtosecond Real‐time observation of wave packet oscillations (Resonance) in dissociation reactions. J Chem Phys. 1988;88:6672–6673.
- Rosker MJ, Rose TS, Zewail AH. Femtosecond Real-time dynamics of photofragment-trapping resonances on dissociative potential energy surfaces. Chem Phys Lett. 1988;146:175–179.
- Johnson PJM, Halpin A, Morizumi T, et al. Local vibrational coherences drive the primary photochemistry of vision. Nat Chem. 2015;7:980–986.
- Schnedermann C, Muders V, Ehrenberg D, et al. Vibronic dynamics of the ultrafast All- trans to 13- cis photoisomerization of retinal in channelrhodopsin-1. J Am Chem Soc. 2016;138:4757–4762.
- Schnedermann C, Liebel M, Kukura P. Mode-specificity of vibrationally coherent internal conversion in rhodopsin during the primary visual event. J Am Chem Soc. 2015;137:2886–2891.
- Wang Q, Schoenlein R, Peteanu L, et al. Vibrationally coherent photochemistry in the femtosecond primary event of vision. Science. 1994;266:422–424.
- Schnedermann C, Yang X, Liebel M, et al. Evidence for a vibrational phase-dependent isotope effect on the photochemistry of vision. Nat Chem. 2018;10:449–455.
- Kahan A, Nahmias O, Friedman N, et al. Following photoinduced dynamics in bacteriorhodopsin with 7-fs impulsive vibrational spectroscopy. J Am Chem Soc. 2007;129:537–546.
- Engel GS, Engel GS, Calhoun TR, et al. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature. 2007;446:782–786.
- Paleček D, Edlund P, Westenhoff S, et al. Quantum coherence as a witness of vibronically hot energy transfer in bacterial reaction center. Sci Adv. 2017;3:e1603141.
- Fang C, Frontiera RR, Tran R, et al. Mapping GFP structure evolution during proton transfer with femtosecond raman spectroscopy. Nature. 2009;462:200–204.
- Fujisawa T, Kuramochi H, Hosoi H, et al. Role of coherent low-frequency motion in excited-state proton transfer of green fluorescent protein studied by time-resolved impulsive stimulated raman spectroscopy. J Am Chem Soc. 2016;138:3942–3945.
- Kuramochi H, Takeuchi S, Yonezawa K, et al. Probing the early stages of photoreception in photoactive yellow protein with ultrafast time-domain raman spectroscopy. Nat Chem. 2017;9:660–666.
- Falke SM, Rozzi CA, Brida D, et al. Coherent ultrafast charge transfer in an organic photovoltaic blend. Science. 2014;344:1001–1005.
- Park M, Neukirch AJ, Reyes-Lillo SE, et al. Excited-state vibrational dynamics toward the polaron in methylammonium lead iodide perovskite. Nat Commun. 2018;9:2525.
- Thouin F, Valverde-Chávez DA, Quarti C, et al. Phonon coherences reveal the polaronic character of excitons in two-dimensional lead halide perovskites. Nat Mater. 2019;18:349–356.
- De Sio A, Troiani F, Maiuri M, et al. Tracking the coherent generation of polaron pairs in conjugated polymers. Nat Commun. 2016;7:13742.
- Batignani G, Fumero G, Srimath Kandada AR, et al. Probing femtosecond lattice displacement upon photo-carrier generation in lead halide perovskite. Nat Commun. 2018;9:1–5.
- Köppel H, Domcke W, Cederbaum LS. Multimode molecular dynamics beyond the Born-oppenheimer approximation. In: Prigogine I, Rice SA, editors. Advance in chemical physics, Vol LVII. Vol. LVII. New York: John Wiley & Sons; 1984. p. 59–246.
- Kühl A, Domcke W. Multilevel redfield description of the dissipative dynamics at conical intersections. J Chem Phys. 2002;116:263.
- Yarkony DR. Conical Intersections: diabolical and often misunderstood. Acc Chem Res. 1998;31:511–518.
- Fink RF, Seibt J, Engel V, et al. Exciton trapping in π-conjugated materials: a Quantum-chemistry-based protocol applied to perylene bisimide dye aggregates. J Am Chem Soc. 2008;130:12858–12859.
- Hoffman DP, Ellis SR, Mathies RA. Characterization of a conical intersection in a charge-transfer dimer with two-dimensional time-resolved stimulated Raman spectroscopy. J Phys Chem A. 2014;118:4955–4965.
- Liebel M, Schnedermann C, Kukura P. Vibrationally coherent crossing and coupling of electronic states during internal conversion in β-carotene. Phys Rev Lett. 2014;112:198302.
- Liebel M, Schnedermann C, Bassolino G, et al. Direct observation of the coherent nuclear response after the absorption of a photon. Phys Rev Lett. 2014;112:238301.
- De Sio A, Sommer E, Nguyen XT, et al. Intermolecular conical intersections in molecular aggregates. Nat. Nanotechnol. 2020;16:63–68.
- Levine BG, Martínez TJ. Isomerization through Conical Intersections. Annu Rev Phys Chem. 2007;58:613–634.
- Takeuchi S, Ruhman S, Tsuneda T, et al. Spectroscopic tracking of structural evolution in ultrafast stilbene photoisomerization. Science. 2008;322:1073–1077.
- Gueye M, Manathunga M, Agathangelou D, et al. engineering the vibrational coherence of vision into a synthetic molecular device. Nat Commun. 2018;9:313.
- Polli D, Weingart O, Brida D, et al. Wavepacket splitting and two-pathway deactivation in the photoexcited visual pigment isorhodopsin. Angew Chemie Int Ed. 2014;53:2504–2507.
- Crespo-Hernández CE, Cohen B, Hare PM, et al. Ultrafast excited-state dynamics in nucleic acids. Chem Rev. 2004;104:1977–2020.
- Keefer D, Schnappinger T, De Vivie-riedle R, et al. visualizing conical intersection passages via vibronic coherence maps generated by stimulated ultrafast X-ray Raman signals. Proc Natl Acad Sci. 2020;117:24069–24075.
- Yonehara T, Hanasaki K, Takatsuka K. Fundamental Approaches to Nonadiabaticity: toward a Chemical Theory beyond the Born–Oppenheimer Paradigm. Chem Rev. 2012;112:499–542.
- Malhado JP, Bearpark MJ, Hynes JT. Non-adiabatic dynamics close to conical intersections and the surface hopping perspective. Front Chem. 2014;2. DOI:https://doi.org/10.3389/fchem.2014.00097.
- Clark J, Nelson T, Tretiak S, et al. Femtosecond torsional relaxation. Nat Phys. 2012;8:225–231.
- Polívka T, Sundström V. Ultrafast dynamics of carotenoid excited states−from solution to natural and artificial systems. Chem Rev. 2004;104:2021–2072.
- Andrzejak M, Skóra T, Petelenz P. Is vibrational coherence a byproduct of singlet exciton fission? J Phys Chem C. 2019;123:91–101.
- Schnedermann C, Alvertis AM, Wende T, et al. A molecular movie of ultrafast singlet fission. Nat Commun. 2019;10:4207.
- Lukman S, Chen K, Hodgkiss JM, et al. Tuning the role of charge-transfer states in intramolecular singlet exciton fission through Side-group engineering. Nat. Commun. 2016;7:13622.
- Schröder FAYN, Turban DHP, Musser AJ, et al. Tensor network simulation of multi-environmental open quantum dynamics via machine learning and entanglement renormalisation. Nat Commun. 2019;10:1062.
- Andrzejak M, Mazur G, Skóra T, et al. Soft selection rules for femtosecond Pump–probe vibrational coherence spectroscopy. J Phys Chem C. 2020;124:23501–23510.
- Stern HL, Cheminal A, Yost SR, et al. Vibronically coherent ultrafast triplet-pair formation and subsequent thermally activated dissociation control efficient endothermic singlet fission. Nat Chem. 2017;9:1205–1212.
- Tamura H, Huix-Rotllant M, Burghardt I, et al. First-principles quantum dynamics of singlet fission: coherent versus thermally activated mechanisms governed by molecular π stacking. Phys Rev Lett. 2015;115:107401.
- Schröder FAYN, Turban DHP, Musser AJ, et al. Tensor network simulation of multi-environmental open quantum dynamics via machine learning and entanglement renormalisation. Nat Commun. 2019;10:1–10.
- Alvertis AM, Lukman S, Hele TJH, et al. Switching between coherent and incoherent singlet fission via solvent-induced symmetry breaking. J Am Chem Soc. 2019;141:17558–17570.
- Ma L, Zhang K, Kloc C, et al. Fluorescence from rubrene single crystals: interplay of singlet fission and energy trapping. Phys Rev B. 2013;87:201203.
- Miyata K, Kurashige Y, Watanabe K, et al. Coherent singlet fission activated by symmetry breaking. Nat Chem. 2017;9:983–989.
- Tamura H, Huix-Rotllant M, Burghardt I, et al. First-principles quantum dynamics of singlet fission: coherent versus thermally activated mechanisms governed by molecular π stacking. Phys Rev Lett. 2015;115:107401.
- Breen I, Tempelaar R, Bizimana LA, et al. Triplet separation drives singlet fission after femtosecond correlated triplet pair production in rubrene. J Am Chem Soc. 2017;139:11745–11751.
- Fuemmeler EG, Sanders SN, Pun AB, et al. A direct mechanism of ultrafast intramolecular singlet fission in pentacene dimers. ACS Cent Sci. 2016;2:316–324.
- Monahan NR, Sun D, Tamura H, et al. Dynamics of the triplet-pair state reveals the likely coexistence of coherent and incoherent singlet fission in crystalline hexacene. Nat Chem. 2017;9:341–346.
- Thampi A, Stern HL, Cheminal A, et al. Elucidation of excitation energy dependent correlated triplet pair formation pathways in an endothermic singlet fission system. J Am Chem Soc. 2018;140:4613–4622.
- Mandal A, Chen M, Foszcz ED, et al. Two-dimensional electronic spectroscopy reveals excitation energy-dependent state mixing during singlet fission in a terrylenediimide dimer. J Am Chem Soc. 2018;140:17907–17914.
- Bossanyi DG, Matthiesen M, Wang S, et al. Emissive spin-0 triplet-pairs are a direct product of triplet–triplet annihilation in pentacene single crystals and anthradithiophene films. Nat Chem. 2021;13:163-171. DOI:https://doi.org/10.1038/s41557-020-00593-y.
- Lukman S, Richter JM, Yang L, et al. Efficient singlet fission and triplet-pair emission in a family of zethrene diradicaloids. J Am Chem Soc. 2017;139:18376–18385.
- Mauck CM, Hartnett PE, Margulies EA, et al. Singlet Fission via an Excimer-Like Intermediate in 3,6-Bis(Thiophen-2-Yl)Diketopyrrolopyrrole derivatives. J Am Chem Soc. 2016;138:11749–11761.
- Hong Y, Kim J, Kim W, et al. Efficient multiexciton state generation in charge-transfer-coupled perylene bisimide dimers via structural control. J Am Chem Soc. 2020;142:7845–7857.
- Busby E, Xia J, Wu Q, et al. Strategy for intramolecular singlet fission mediated by charge-transfer states in donor–acceptor organic materials. Nat Mater. 2015;14:426–433.
- Hu J, Xu K, Shen L, et al. New insights into the design of conjugated polymers for intramolecular singlet fission. Nat Commun. 2018;9:2999.
- Stern HL, Musser AJ, Gelinas S, et al. Identification of a triplet pair intermediate in singlet exciton fission in solution. Proc Natl Acad Sci. 2015;112:7656–7661.
- Dover CB, Gallaher JK, Frazer L, et al. Endothermic singlet fission is hindered by excimer formation. Nat Chem. 2018;10:305–310.
- JDirect Determination of the S1 Excited-State Energies of Xanthophylls by Low-Temperature Fluorescence Spectroscopy. J Phys Chem A. 2002;106:4815-4824.
- Petek H, Bell AJ, Choi YS, et al. The 2 1Ag State of trans,trans-1,3,5,7-octatetraene in free jet expansions. J Chem Phys. 1993;98:3777.
- Albrecht AC. On the theory of raman intensities. J Chem Phys. 1961;34:1476–1484.
- Tao S, Ohtani N, Uchida R, et al. Relaxation dynamics of photoexcited excitons in rubrene single crystals using femtosecond absorption spectroscopy. Phys Rev Lett. 2012;109:097403.
- Ma L, Galstyan G, Zhang K, et al. Two-photon-induced singlet fission in rubrene single crystal. J Chem Phys. 2013;138:184508.
- Ward KA, Richman BR, Biaggio I. nanosecond pump and probe observation of bimolecular exciton effects in rubrene single crystals. Appl Phys Lett. 2015;106:223302.
- Gieseking B, Schmeiler T, Müller B, et al. Effects of characteristic length scales on the exciton dynamics in rubrene single crystals. Phys Rev B. 2014;90:205305.
- Finton DM, Wolf EA, Zoutenbier VS, et al. Routes to singlet exciton fission in rubrene crystals and amorphous films. AIP Adv. 2019;9:9.
- Deng GH, Wei Q, Han J, et al. Vibronic fingerprint of singlet fission in hexacene. J Chem Phys. 2019;151:054703.
- Bera K, Douglas CJ, Frontiera RR. Femtosecond Raman microscopy reveals structural dynamics leading to triplet separation in rubrene singlet Fission. J Phys Chem Lett. 2017;8:5929–5934.
- Bera K, Kwang SY, Frontiera RR. Advances in singlet fission chromophore design enabled by vibrational spectroscopies. J Phys Chem C. 2020;1. DOI:https://doi.org/10.1021/acs.jpcc.0c06725
- Fumero G, Schnedermann C, Batignani G, et al. Two-dimensional impulsively stimulated resonant Raman spectroscopy of molecular excited-states. Phys Rev X. 2020;10:1–26.
- Kuramochi H, Takeuchi S, Kamikubo H, et al. Fifth-order Time-domain Raman spectroscopy of photoactive yellow protein for visualizing vibrational coupling in its excited state. Sci Adv. 2019;5:eaau4490.
- Xue L, Song X, Feng Y, et al. General dual-switched dynamic singlet fission channels in solvents governed jointly by chromophore structural dynamics and solvent impact: singlet prefission energetics analyses. J Am Chem Soc. 2020;142:17469–17479.
- Montero R, Martínez-Martínez V, Longarte A, et al. Singlet fission mediated photophysics of BODIPY dimers. J Phys Chem Lett. 2018;9:641–646.
- Andrzejak M, Skóra T, Petelenz P. Limitations of generic chromophore concept for femtosecond vibrational coherences. J Phys Chem C. 2020;124:3529–3535.
- Sciaini G, Miller RJD. Femtosecond electron diffraction: heralding the era of atomically resolved dynamics. Reports Prog Phys. 2011;74:096101.
- Seiler H, Krynski M, Zahn D, et al. Nuclear dynamics of singlet exciton fission: a direct observation in pentacene single crystals. 2020.
- Costantini R, Faber R, Cossaro A, et al. Picosecond timescale tracking of pentacene triplet excitons with chemical sensitivity. Commun Phys. 2019;2:56.
- Kim JG, Nozawa S, Kim H, et al. Mapping the emergence of molecular vibrations mediating bond formation. Nature. 2020;582:520–524.