
Abstract
In this study, the complete mitochondrial genome of plant pathogenic fungus, Mycosphaerella pinodes, was sequenced. The nucleotide composition of the genome is: 36.0% of A, 15.0% of C, 14.6% of G and 34.5% of T. The mitochondrial genome is 55 973 bp in length and consists of 11 protein-coding genes, two ribosomal RNAs and 25 tRNA genes. The mitogenome analysis of M. pinodes provide a molecular basis for further studies on molecular systematics and evolutionary dynamics of Ascomycetes fungi especially belonging to Dothideomycetes.
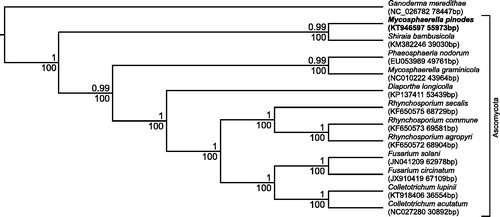
Ascochyta blight is one of the most devastating diseases of field pea worldwide, and has been reported in many major pea-growing areas, including Poland (Boros & Marcinkowska Citation2010). Among the fungi, causing ascochyta blight symptoms, a hemibiotrophic fungus Mycosphaerella pinodes is the most damaging pathogen. The strain of M. pinodes sequenced in this study (GenBank accession number: KT946597) was isolated from pea leaves of cv. Piast with ascochyta blight symptoms in Tomaszkowo near Olsztyn, Poland (53°42'44.1”N + 20°25'55.3”E) (Okorski & Majchrzak Citation2009) and has been deposited and stored in the Department of Entomology, Phytopathology and Molecular Diagnostics Fungal Culture Collection (Accession No 165/T) (University of Warmia and Mazury in Olsztyn, Poland). The DNA from a single spore of a 14-d 165/T M. pinodes culture was extracted with DNeasy Plant Mini DNA Extraction Kits (Qiagen Inc., Valencia, CA). The genome libraries were prepared using a Nextera XT kit (Illumina Inc., San Diego, CA) from genomic DNA. The prepared libraries were sequenced on the Illumina MiSeq platform (Illumina Inc.) with the 300 bp paired-end read, version 3. The trimmed reads were mapped to the reference mitogenomes of M. graminicola (NC_010222, EU090238), Fusarium solani (NC_016680), Rhynchosporium agropyri (NC_023125) and Phaeosphaeria nodorum (NC_009746) using Geneious Mapper (version 8.0.4) (http://www.geneious.com, Kearse et al. Citation2012). All contigs, containing annotated genes, were remapped to elongate them. Obtained scaffolds were assembled to full-length mitogenome and closed to circular mtDNA. The mitogenome was annotated using PlusMapper (Dong et al. Citation2004) and was manually adjusted. ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to verify annotated genes and to identify any potential genes in uncoding regions. The rRNA subunits were identified by aligning to numerous fungi reference sequences using Megablast (Morgulis et al. Citation2008). The M. pinodes mitogenome structure is complexed and its different regions are similar to mitogenomes of a few different organisms (but always among fungi). That is why we used four main different reference genomes and much more supported reference. For instance, the ‘cox1 & 2’ gene construction, where cox1 and cox2 subunits are coding by one gene is similar to P. nodorum (NC_009746) ‘cox1 & 2’ gene, but intron–exon structure of this gene is similar as on the R. agropyri (NC_023125) mitogenome. The full length of the M. pinodes mitogenome is 55 973 bp. We identified 38 genes: 11 protein-coding genes, two ribosomal RNAs and 25 tRNA genes (for all 20 amino acids). The phylogenetic relations of sequenced mitogenome of M. pinodes among Ascomycota is presented in . High bootstrap and posterior probability values show that presented relations are stable.
Figure 1. The phylogenetic tree of the Mycosphaerella pinodes mitogenome and related organisms, members of Phylum: Ascomycota and Ganoderma meredithae (the member of Phylum: Basidiomycota) as the root branch. The full mitochondrial DNA of Mycosphaerella pinodes (KT946597 55 973 bp), Mycosphaerella graminicola (NC010222 43 964 bp), Phaeosphaeria nodorum (EU053989 49 761 bp), Shiraia bambusicola (KM382246 39 030 bp), Diaporthe longicolla (KP137411 53 439 bp), Fusarium circinatum (JX910419 67 109 bp), Fusarium solani (JN041209 62 978 bp), Colletotrichum lupini (KT918406 36 554 bp), Colletotrichum acutatum (NC027280 30 892 bp), Rhynchosporium secalis (KF650575 68 729 bp), Rhynchosporium agropyri (KF650572 68 904 bp), Rhynchosporium commune (KF650573 69 581 bp), Ganoderma meredithae (NC_026782 78 447 bp) were aligned using MAFT algorithm on: Multiple Sequence Alignment Software Version 7. The phylogenetic analysis were done using the maximum-likelihood method based on the GTR + G substitution model in RAxML version 8.0.26 tool (Stamatakis Citation2014) and Bayes method in BEAST version 1.8.1 tool (Drummond et al. Citation2012). The percentage of trees in which the associated taxa clustered together (for 1000 bootstrap replications) is shown below the node and the posterior probability value is shown above the node.
Declaration of interest
This study was funded by the Polish Ministry of Agriculture and Rural Development under the project No. HOR. 3.6. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Boros L, Marcinkowska J. 2010. Assessment of selected pea genotypes reaction to ascochyta blight under field conditions and the impact of disease severity on yield components. Can J Agric Sci. 2:84–91.
- Dong X, Stothard P, Forsythe IJ, Wishart DS. 2004. PlasMapper: a web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 32:W660–W664.
- Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:647–1649.
- Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schäffer AA. 2008. Database indexing for production MegaBLAST searches. Bioinformatics 15:1757–1764.
- Okorski A, Majchrzak B. 2009. Fungi isolated from infected tissues of pea after applying biological control product EM 1. Prog Plant Prot/Postępy w Ochronie Roślin 49:242–245 (in Polish).
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313.