
Abstract
The complete mitochondrial genome of Halcyon smyrnensis was determined in this study. The mitogenomic length of Halcyon smyrnensis was 17 318 bp, including 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes and one control region. Phylogenetic analyses show that H. leucocephala, H. pileata and H. smyrnensis congregated together, and our sample was sister to H. pileata.
Halcyon smyrnensis is a tree kingfisher, widely distributed in Asia from Turkey east through the India subcontinent to the Philippines. Halcyon smyrnensis has been listed as a class least concern (LC) species in IUCN (Citation2012) Red List of Threatened Species. We determined the complete mitochondrial genome of H. smyrnensis, and should contribute to researches of mitogenomic evolution, phylogenetic relationship and conservation genetics in Halcyon (Moritz Citation1994; Bayer et al. Citation2012; Qu et al. Citation2015; Wang et al. Citation2015).
The muscle sample from H. smyrnensis (voucher YTSM-HP1M, stored in the marine ecology lab at the College Life Science, Yantai University) was collected in Yacheng Town (E 109.18°–N 18.36°), Sanya City, Hainan Island, China. By polymerase chain reaction, 22 primer pairs were used to acquire the mitochondrial genome of H. pileata in this study. The complete mtDNA of H. pileata (17 318 bp in length, GenBank accession no. KT965614) had 13 protein-coding genes, 22 tRNA genes, two rRNA genes (12 SrRNA and 16 SrRNA) and one control region (CR, D-loop gene). Twenty-nine genes in H. pileata were distributed on the H-strand, and nine genes were distributed on the L-strand. Overall base composition of the mitogenome is 33.76% A, 22.88% T, 32.72% C and 13.63% G.
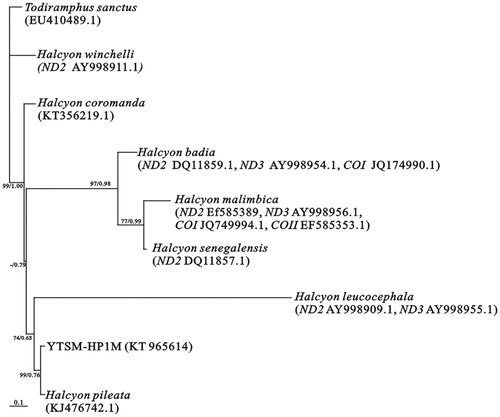
The phylogenetic position of H. pileata was estimated using maximum-likelihood (ML) and Bayesian inference (BI) (). ML analyses produced the similar topology as BI tree. RAxML v 7.2.6 (Stamatakis Citation2006) was used to conduct ML tree with the best model (GTR + I + G, 10 randomized starting trees). Bayesian phylogenetic analyses were performed using MRBAYES version 3.1.2 (Huelsenbeck & Ronquist Citation2001) with the best model (GTR + I + G), and four simultaneous Monte Carlo Markov chains (MCMCs) were run for 5 × 106 generations, sampling a tree every 100 generations. ML analyses produced the similar topology as the BI tree. The results show that H. leucocephala, H. pileata and H. smyrnensis congregated together, and our sample was sister to H. pileata ().
Figure 1. Maximum-likelihood and Bayesian trees based on the GTR + I + G model of eight species in Halcyon using partial available genes. The phylogenetic tree was rooted using Todiramphus sanctus. Our sample was YTSM-HP1M. Values of bootstrap percentage were computed in ML trees (first numbers) and posterior probability (second numbers) in BI trees were shown for the key node more than 0.60. ‘‘–” mean that the bootstrap value was less than 50%. The GenBank accession number of the mitogenome of each species was listed following the species name.
Declaration of interest
The authors report that they have no conflict of interest. This study was supported by National Natural Science Foundation of China (31460562 to Jiangyong Qu), Natural Science Foundation of Shandong province (ZR2013DM002 to Chenghua Guo) and Co-operation Fund between University and Sanya City (2014YD37 to Jiangyong Qu).
References
- Bayer CSO, Sackman AM, Bezold K, Cabe PR, Marsh DM. 2012. Conservation genetics of an endemic mountaintop salamander with an extremely limited range. Conserv Genet. 13:443–454.
- Huelsenbeck JP, Ronquist F. 2001. mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755.
- IUCN. 2012. The International Union for Conservation of Nature (IUCN) Red List of Threatened Species. Version 2013. 2. International Union for Conservation of Nature; [cited 2013 Nov 26].
- Moritz C. 1994. Application of mitochondrial DNA analysis in conservation: a critical review. Mol Ecol. 3:401–411.
- Qu JY, Cong JS, Guo CH, Hou JH, Zhen JR, Shi BY. 2015. Complete mitochondrial genome of the endangered greater coucal Centropus sinensis (Cuculiformes: Centropidae: Centropus). Mitochondrial DNA, doi: 10.3109/19401736.2015.1118092.
- Stamatakis A. 2006. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690.
- Wang N, Liang B, Huo J, Liang W. 2015. Complete mitochondrial genome and the phylogenetic position of the Lesser Cuckoo, Cuculus poliocephalus (Aves: Cuculiformes). Mitochondrial DNA, doi:10.3109/19401736.2015.1089547.