
Abstract
The complete mitochondrial genome of Gyrodactylus kobayashii was 14 786 bp in length, containing 12 protein-coding genes (lacking Atp8), 22 tRNA genes, two rRNA genes and two major non-coding regions (NC1 and NC2). The overall A + T content of mitochondrial genome was 71.6%. A close relationship between G. kobayashii and the three Gyrodactylus species (G. salaris, G. thymalli and G. derjavinoides) was uncovered in the phylogenetic tree based on amino acid sequences.
Gyrodactylus kobayashii was the most common Gyrodactylus species on the fins and gills of goldfish Carassius auratus. The worm was collected on goldfish from Wuhan (30°31'23”N, 114°23'01”E), China, and was identified by morphology and ITS molecular marker (Li et al. Citation2013).
The complete mitochondrial genome of G. kobayashii (GenBank accession no. KU057942) was sequenced by using long PCR and Sanger method of DNA sequencing. The circular mitogenome was 14 786 bp long and contained 12 protein-coding genes (PCGs, lacking Atp8), 22 tRNA genes, two rRNA genes and two major non-coding regions (NC1 and NC2) (). All the genes were transcribed from the same strand. The base composition was 41.9% T, 11.1% C, 29.7% A and 17.3% G. The disproportionally overall A + T content was 71.6%, which was higher than any of the three Gyrodactylus species (G. salaris, 62.3%; G. thymalli 62.8% and G. derjavinoides, 68.2%) (Huyse et al. Citation2007; Plaisance et al. Citation2007; Huyse et al. Citation2008). The gene order of G. kobayashii matched exactly with the three Gyrodactylus species.
The length of 12 PCGs was 9945 bp, with 71.6% A + T content. ATG was the unique start codon. Nad5, Nad3, Nad2, Nad1 and Atp6 appeared to use TAG as stop codon, whereas the rest of the PCGs used the stop codon TAA, and no premature stop codon (TA or T) was found. Total length of the 22 tRNA genes was 1436 bp, varying from 58 bp (tRNASer(AGN)) to 72 bp (tRNAGlu and tRNAAla). All tRNAs could be fold into the conventional secondary structure, except for three unorthodox tRNAs, tRNASer(AGN), tRNASer(UCN) and tRNACys lacked DHU arms. The rrnL and rrnS were 955 bp and 707 bp in size, respectively. They were flanked by tRNAThr and Cox2, and separated by tRNACys, as demonstrated in the monopisthocotyleans (Huyse et al. Citation2007; Plaisance et al. Citation2007; Huyse et al. Citation2008; Perkins et al. Citation2010; Ye et al. Citation2014; Zhang et al. Citation2014a,Citationb).
There were five cases of overlapping regions within the mitogenome. The overlap between Nad4L and Nad4 was common in metazoan mtDNAs (von Nickisch-Rosenegk et al. Citation2001), with the exception of Benedenia hoshinai and B. seriolae (Perkins et al. Citation2010). There were 21 short intergenic regions ranging from 1 bp to 111 bp. In addition, the two long non-coding regions, NC1 (between tRNAPhe and Atp6) and NC2 (between tRNAMet and tRNASer(UCN)) were 778 bp and 783 bp long, 67.0% and 68.2% for the AT content, respectively. The high similarity over 686 bp sequences was found between NC1 and NC2, with the differences in eight substitutions and three indels.
The phylogenetic analysis was performed with 2858 homologous concatenated amino acid sequences representing 12 protein-coding genes from nine available mitochondrial genomes and G. kobayashii mitogenome (this study), implementing maximum-likelihood (ML) and Bayesian inference (BI) analyses. Both the phylogenetic methods produced the same tree topology in the branching patterns. Gyrodactylus kobayashii was closely relate to the three Gyrodactylus species with extremely high bootstrap resampling (ML) and posterior probability (BI) values ().
Figure 1. Phylogenetic tree of Gyrodactylus kobayashii and selected monopisthocotyleans based on the concatenated amino acids representing 12 mitochondrial protein-coding genes. The MtZoa model for maximum-likelihood analysis and MtREV model for Bayes analysis are selected according to AIC criterion. Scale bar represents the estimated number of substitutions per site. The numbers at the nodes indicate posterior probability (upper value) and bootstrap probability (lower value).
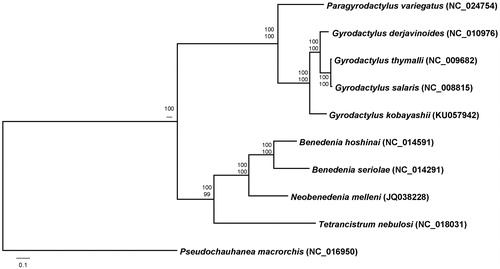
Table 1. Organization of the mitochondrial genome of Gyrodactylus kobayashii.
Funding information
This work was supported by the Earmarked Fund for China Agriculture Research System (CARS-46-08), the National Natural Science Foundation of China (31272695 and 31572658) and the major scientific and technological innovation project of Hubei Province (2015ABA045).
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the manuscript.
References
- Huyse T, Buchmann K, Littlewood DTJ. 2008. The mitochondrial genome of Gyrodactylus derjavinoides (Platyhelminthes: Monogenea) – a mitogenomic approach for Gyrodactylus species and strain identification. Gene. 417:27–34.
- Huyse T, Plaisance L, Webster BL, Mo TA, Bakke TA, Bachmann L, Littlewood DTJ. 2007. The mitochondrial genome of Gyrodactylus salaris (Platyhelminthes: Monogenea), a pathogen of Atlantic salmon (Salmo salar). Parasitology. 134:739–747.
- Li RR, Li WX, Wu XD, Wang GT. 2013. Identification of Gyrodactylus species in goldfish (Carassius auratus) through morphological study and the analysis of the rDNA ITS sequence. Acta Hydrobiol Sin. 38:903–909.
- Perkins EM, Donnellan SC, Bertozzi T, Whittington ID. 2010. Closing the mitochondrial circle on paraphyly of the Monogenea (Platyhelminthes) infers evolution in the diet of parasitic flatworms. Int J Parasitol. 40:1237–1245.
- Plaisance L, Huyse T, Littlewood DTJ, Bakke TA, Bachmann L. 2007. The complete mitochondrial DNA sequence of the monogenean Gyrodactylus thymalli (Platyhelminthes: Monogenea), a parasite of grayling (Thymallus thymallus). Mol Biochem Parasitol. 154:190–194.
- von Nickisch-Rosenegk M, Brown WM, Boore JL. 2001. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that Platyhelminths are Eutrochozoans. Mol Biol Evol. 18:721–730.
- Ye F, King SD, Cone DK, You P. 2014. The mitochondrial genome of Paragyrodactylus variegatus (Platyhelminthes: Monogenea): differences in major non-coding region and gene order compared to Gyrodactylus. Parasit Vectors. 7:377.
- Zhang J, Wu X, Li Y, Xie M, Li A. 2014a. The complete mitochondrial genome of Tetrancistrum nebulosi (Monogenea: Ancyrocephalidae). Mitochondrial DNA. 1736:1–2.
- Zhang J, Wu X, Li Y, Zhao M, Xie M, Li A. 2014b. The complete mitochondrial genome of Neobenedenia melleni (Platyhelminthes: Monogenea): mitochondrial gene content, arrangement and composition compared with two Benedenia species. Mol Biol Rep. 41:6583–6589.