
Abstract
In this study, the near complete mitogenome sequence (15,469 bp) of Oreolalax major was determined using polymerase chain reaction (PCR). It includes 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes and 19 transfer RNA (tRNA) genes (GenBank accession number KU310894). The features of O. major have one more tRNA gene (tRNAMet) behind the original one before ND2 which is similar to Leptobrachium boringii. Phylogenetic analyses were based on the concatenated sequences of the 13 protein-encoding genes of O. major and other related species.
The Large toothed toad, Oreolalax major (Liu & Hu 1960) belongs to family Megophryidae, genus Oreolalax. It is an endemic species of China, which is distributed in Hengduan Mountains of western Sichuan and southern Gansu Provinces (Fei et al. Citation2010; Frost Citation2015). But there is no mitogenome sequence of genus Oreolalax published until now. In this study, we sequence the mitogenome of O. major in order to explore the relationship among most of the genera of Megophryidae.
The O. major sample (Specimen voucher: 20120256) was collected from Wawushan Nature Reserve, Hongya (29°34′26.72″N, 102°56′35.73″E., elev. 1394m), Sichuan, China in June, 2015. It was fixed in 75% ethanol and deposited in the Museum of Sichuan Agricultural University. The total DNA was extracted and purified from muscle tissue using the Ezup pillar genomic DNA extraction kit (Sangon Biotech, Shanghai, China). We used twenty-three pairs of polymerase chain reaction (PCR) primers published by Zhang et al. (Citation2013) and Kurabayashi & Sumida. (Citation2009) to amplification and sequencing the mitogenome, internal primers were also designed when above-mentioned primers did not work. The neighbour-joining (NJ) tree based on 13 protein-coding genes (PCGs) of mitochondrial genome was reconstructed by MEGA 6.0 using Tamura–Nei model (Tamura et al. Citation2013).
The near complete mitochondrial genome of O. major is 15,469 bp in length and includes 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes and 19 transfer RNA (tRNA) genes (GenBank accession number KU310894). The features of O. major have one more tRNA gene (tRNAMet) behind the original one before ND2 which is similar to Leptobrachium boringii (Xu et al. Citation2016) and most genes are encoded on H-strand except ND6, tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer(UCN) and tRNAGlu genes. We regrettably did not sequence the region between 12S rRNA and Cyt b genes which contains 4 rRNA genes and 1 D-loop region because of difficult to amplify. There are two types of initiation codons used for 13 PCGs and most PCGs initiate from ATG, while others (COI, ND5, ND6) start with GTG. In addition, 5 PCGs (ND1, ATP6, COIII, ND4, Cyt b) terminate in incomplete stop codon T, the same number of PCGs (ND2, COII, ATP8, ND3, ND4L) use complete stop codon TAA, and the rest of PCGs end with AGG. The 12S rRNA and 16S rRNA are determined to be 936 bp and 1598 bp long respectively and all the tRNA genes can constitute typical cloverleaf secondary structure exclude tRNASer(AGY).
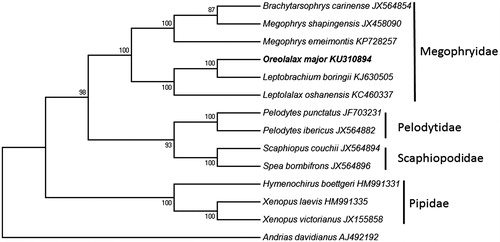
The NJ analysis of the combined data set indicates that all samples of Megophryidae form a strong supported (bootstrap value = 100) monophyletic group () and all families (e.g. Pelodytidae/Scaphiopodidae/Pipidae) included in the analysis are monophyletic. The mitogenomic phylogenetic tree is largely consistent with the previous molecular results (Zhang et al. Citation2013).
Figure 1. Neighbour-joining (NJ) tree was built on mitogenomic sequences of all 13 combined protein-coding genes. Andrias davidianus was used as outgroup. The NJ tree was reconstructed with the Tamura–Nei model, and the numbers on branches are NJ bootstrap values for 1000 replicates.
Funding information
This research was supported by the National Natural Science Foundation of China to M.W. Zhang (31470444), Foundation of Education Department of Sichuan to M.W. Zhang (13ZA0262), and the Innovative Research Team in University of Sichuan Bureau of Education (14TD0002).
Acknowledgements
We are grateful to the Project supporting by Sichuan Wawushan Nature Reserve Multi-disciplinary Scientific Expedition.
Disclosure statement
The authors report no conflicts of interest.
Related Research Data
References
- Fei L, Ye CY, Jiang JP. 2010. Colored atlas of Chinese amphibian. Chengdu (China): Sichuan Science and Technology Press.
- Frost DR. [Internet]. 2015. Amphibian species of the world: an online reference. Version 6. Electronic database. American Museum of Natural History, New York, USA. [cited 2015 Dec 30]. Available from: http://research.amnh.org/herpetology/amphibia/index.html
- Kurabayashi A, Sumida M. 2009. PCR primers for the neobatrachian mitochondrial genome. Curr Herpetolo. 28:1–11.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729.
- Xu Q, Liu S, Wan R, Yue B, Zhang X. 2016. The complete mitochondrial genome of Vibrissaphora boringii (Anura: Megophryidae). Mitochondrial DNA. 27:758–759.
- Zhang P, Liang D, Mao RL, Hillis DM, Wake DB, Cannatella DC. 2013. Efficient sequencing of anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol Boil E. 30:1899–1915.