
Abstract
The following report details the first annotated mitochondrial genome for the Carmagnola variety of Cannabis sativa, the first reference genome for the Cannabaceae family. The total length is 415,499 bp and contains 54 genes, which sub-divide into 38 protein-coding genes, 15 tRNA genes, and 3 rRNA genes.
Keywords:
Cannabis sativa has been an important agricultural crop throughout human history. Despite its multiple uses, C. sativa has been classified as a drug in many countries, a political aspect that has impeded scientific investigation on the plant (Li Citation1973; Russo Citation2007). C. sativa belongs to the family Cannabaceae, which is composed of 10 different genera and ∼100 species (Bell et al. Citation2010). The C. sativa plants are wind-pollinated, usually annual and most individuals are dioecious, but monoecious individuals occur (Li Citation1973). In addition to the critical functions of the mitochondrion, its genome can reveal important evolutionary patterns. Further, there are a disproportionately low number of assembled plant mtDNA genomes on NCBI due, in part, to inherent difficulties in assembly. Thus, the availability of a complete assembled, and annotated C. sativa mitochondrial genome provides a useful tool for a variety of studies in diverse biological fields.
DNA from a female plant from the Carmagnola variety of C. sativa grown in Colorado (39°58’47.90” N, 105°4’30.05” W) was extracted using a Quiagen DNeasy plant kit (Germantown, MD). The whole-genome library of genomic DNA was sequenced using the Illumina Hiseq2500 platform (San Diego, CA) and yielded 40,963,006, 130bp paired-end reads with an average insert size of 150bp. The published Cannabis genome assembly includes a partially assembled mitochondrion of the Purple Kush variety (van Bakel et al. Citation2011), which we downloaded from the short-read archives of Genbank and used as a reference to generate an improved, error-corrected, and annotated Cannabis mitochondrial genome. The mitochondrion was then assembled denovo using SPAdes genome assembler v.3.1.1 (Bankevitch et al. Citation2012). We merged the contigs that overlapped, filled remaining gaps, and extended the end of the assembled contig until we had a circularized sequence resulting in the complete genome. Subsequently, we performed error correction by aligning the trimmed reads to the draft genome. The 54 genes present in our genome were annotated using Mitofy (Alverson et al. Citation2010).
We downloaded 17 genes (atp1, atp6, atp9, cox1, cox2, cox3, matR, nad1, nad2, nad4, nad4L, nad5, nad6, nad7, nad9, rps3, and rps12) shared among 11 species from the public repository NCBI (Table 1). These CDS (coding sequences) were aligned using the software Clustal X with default parameters (Larkin et al. Citation2007). The 17 alignments were concatenated and a maximum likelihood tree () was constructed using MEGA v. 6.0612 (Takamura et al. Citation2013). The alignment of these 17 CDS from the 11 species was ∼16,173 bp.
Figure 1. Maximum likelihood tree. We used 17-shared genes from nine species from the subclass Rosids: Order Brassicales – Arabidopsis thaliana (NC_001284), Brassica rapa (NC_016125), and Carica papaya (NC_012116); Order Cucurbitales – Cucurbita pepo (NC_014050); Order Fabales – Glycine max (NC_020455), and Millettia pinnata (NC_016742); Order Malpighiales – Ricinus communis (NC_015141); and Order Rosales -–Malus domestica (NC_018554), and Cannabis sativa (KR_059940). We used two species from the subclass Asterids and order Lamiales as outgroups: Ajuga reptans (NC_023103), and Mimulus guttatus (NC_018041).
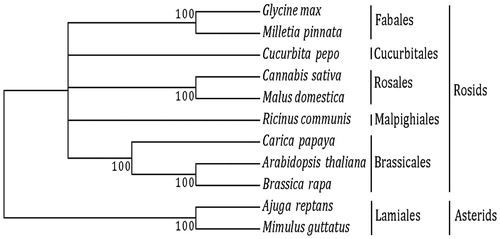
This phylogenetic analysis illustrates the relationship between the species within each order. Once we collapse, the low bootstrap orders into polytomies, our phylogenetic tree reflects the most accepted relationship between the orders in the angiosperms. Using zPicture10 (Ovcharenko et al. Citation2004), we aligned our assembled genome to the un-annotated Purple Kush13 and LA Confidential (http://www.medicinalgenomics.com/chloroplast-and-mitochondrial-haplotypes-of-cannabis) genomes.
Our fully error-corrected assembly differs from both, with 69 mismatches and 271 inserted/deleted bases relative to Purple Kush; or 164 mismatches and 212 inserted/deleted bases relative to LA Confidential. As our genome assembly and error correction algorithms differ, it is not clear if these discrepancies represent biological distinctions or instead errors in sequencing or assembly. We are confident that our assembly, KR059940, represents the best possible one given in the current technology.
Acknowledgements
We would like to thank Ben Holmes of Centennial Seeds for providing the DNA, support, and help with this project. We also thank Erin Collier-Zans for assembly advice and support.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content.
Funding
This project was supported by the University of Colorado Foundation gift fund [13401977-Fin8], the University of Colorado’s Innovative Seed Grant Program grant to N. K., and the NSF IGERT Fellowship for Interdisciplinary Quantitative Biology (IQBiology) to K. G. K.
References
- Alverson, et al. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27:1436–1448.
- Bankevitch, et al. 2012. SPAdes: a new genome assembly algorithm and its application to single-cell sequencing. J Comput Biol. 19:455–477.
- Bell CD, et al. 2010. The age and diversification of the angiosperms re-revisited. Am J Bot. 97:1296–1303.
- Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics. 23:2947–2948.
- Li HL. 1973. An archaeological and historical account of cannabis in China. Econ Bot. 28:437.
- Ovcharenko I, et al. 2004. zPicture: dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 14:472–477.
- Russo EB. 2007. History of cannabis and its preparations in saga, science, and sobriquet. Chem Biodivers. 4:1614–1648.
- Takamura K, et al. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729.
- van Bakel, et al. 2011. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12:R102.